Download as PPSX, PPTX



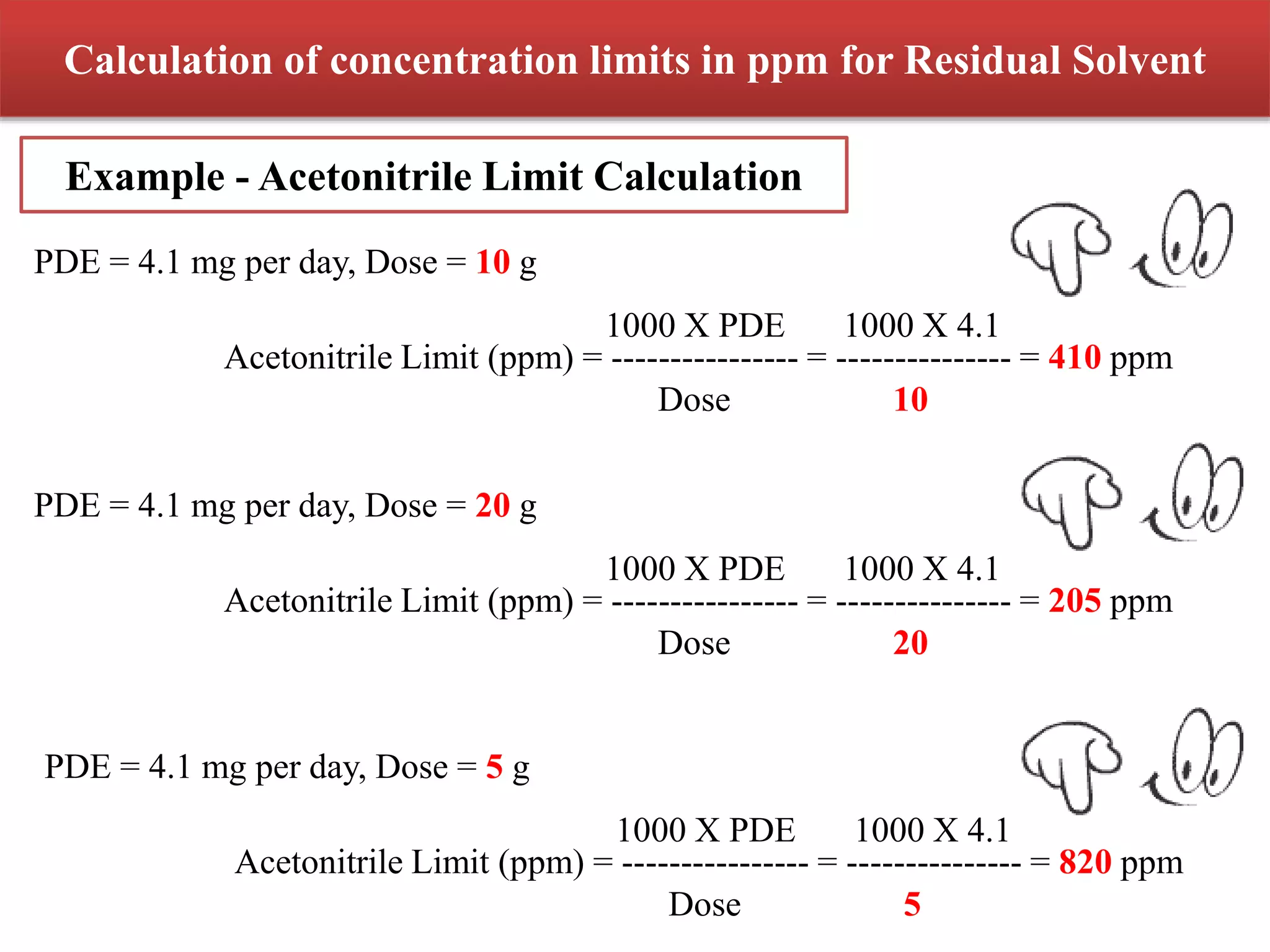

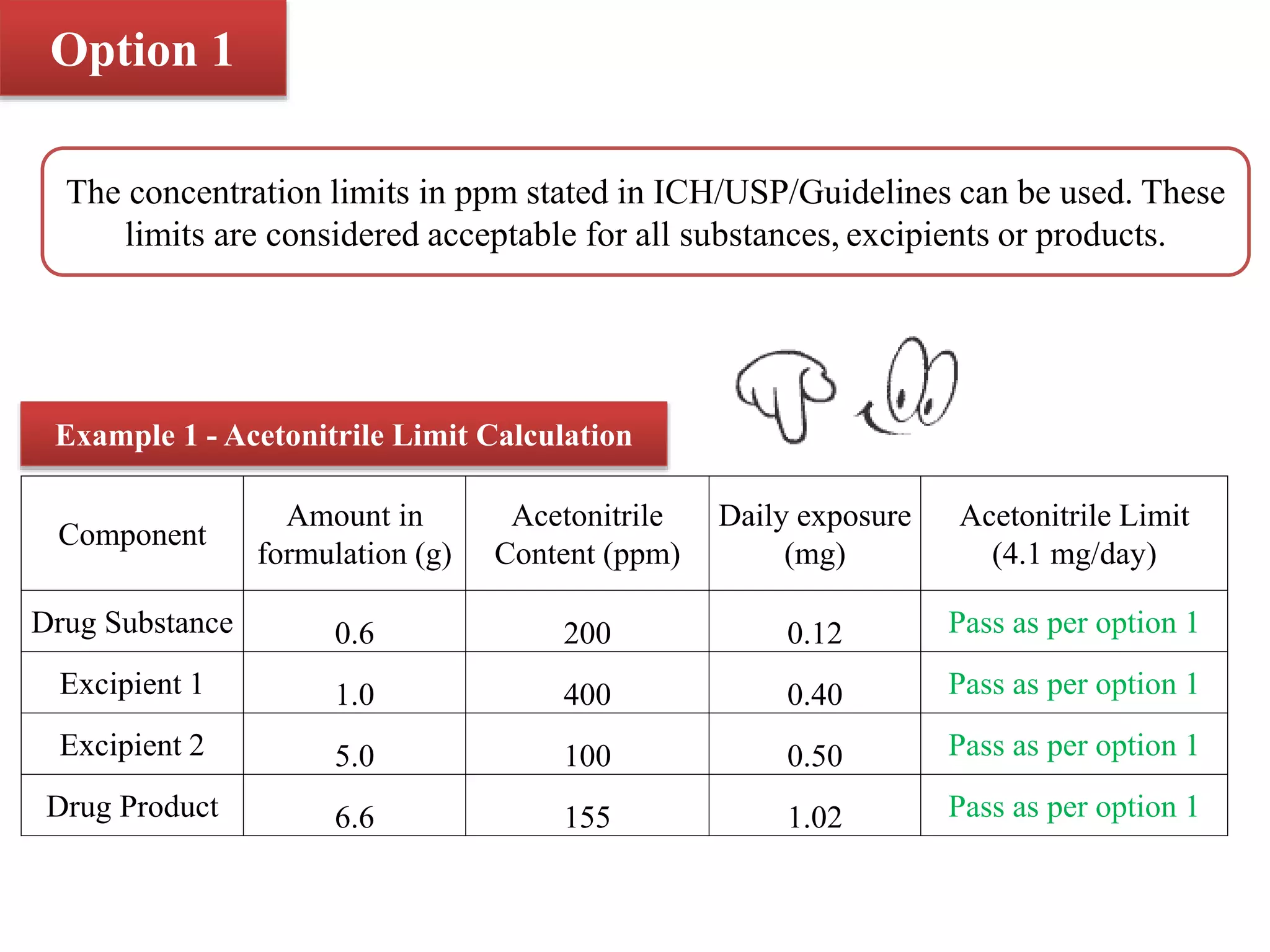


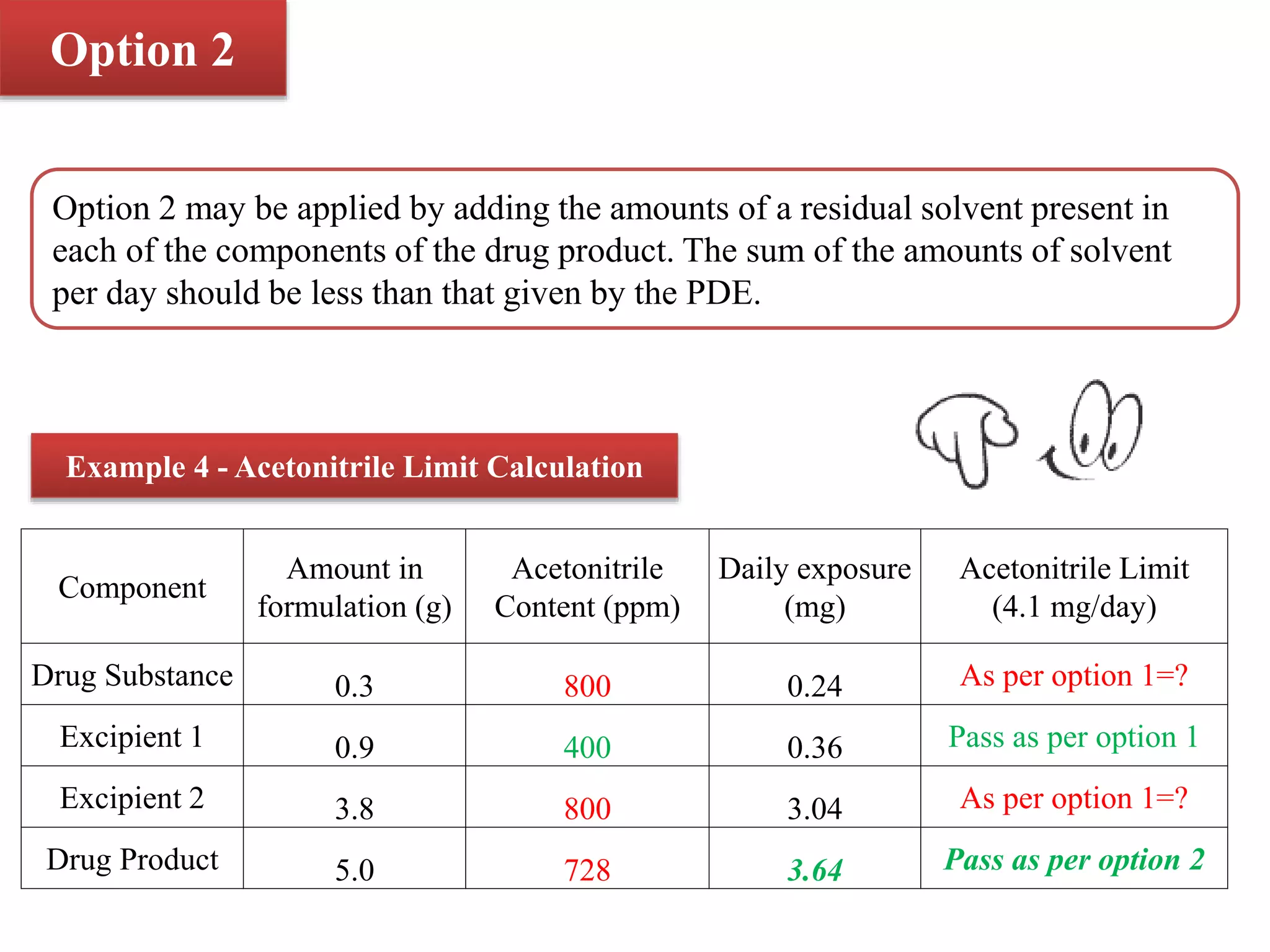
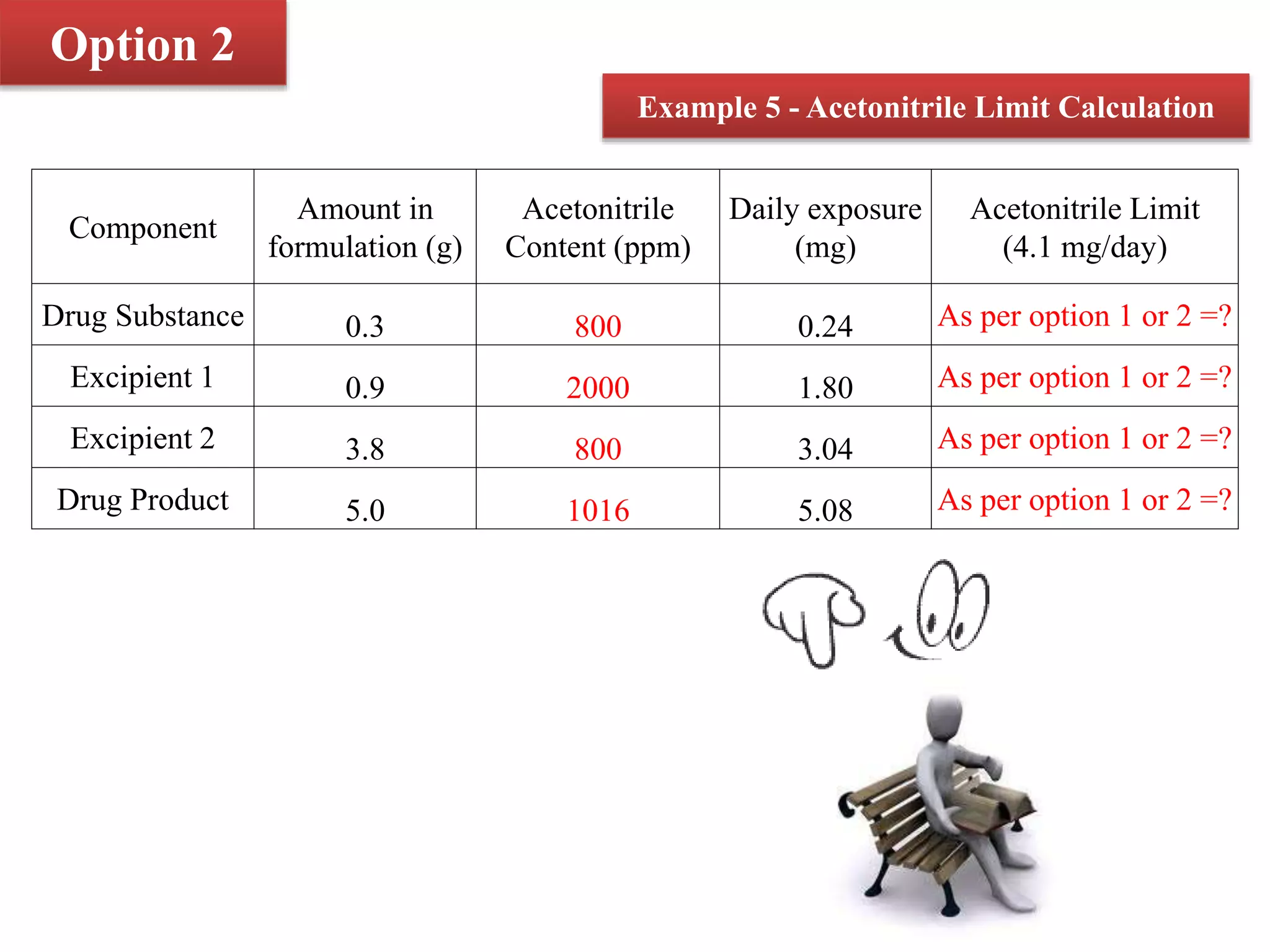



The document discusses two options for describing limits of Class 2 solvents such as acetonitrile in pharmaceutical products. Option 1 uses set concentration limits from guidelines that are acceptable if the daily dose is below 10g. Option 2 involves calculating the total daily exposure of a solvent by summing the amounts from all components, which must be below the permitted daily exposure level. An example shows how the limits are calculated and applied to determine if a product meets the requirements.






















































