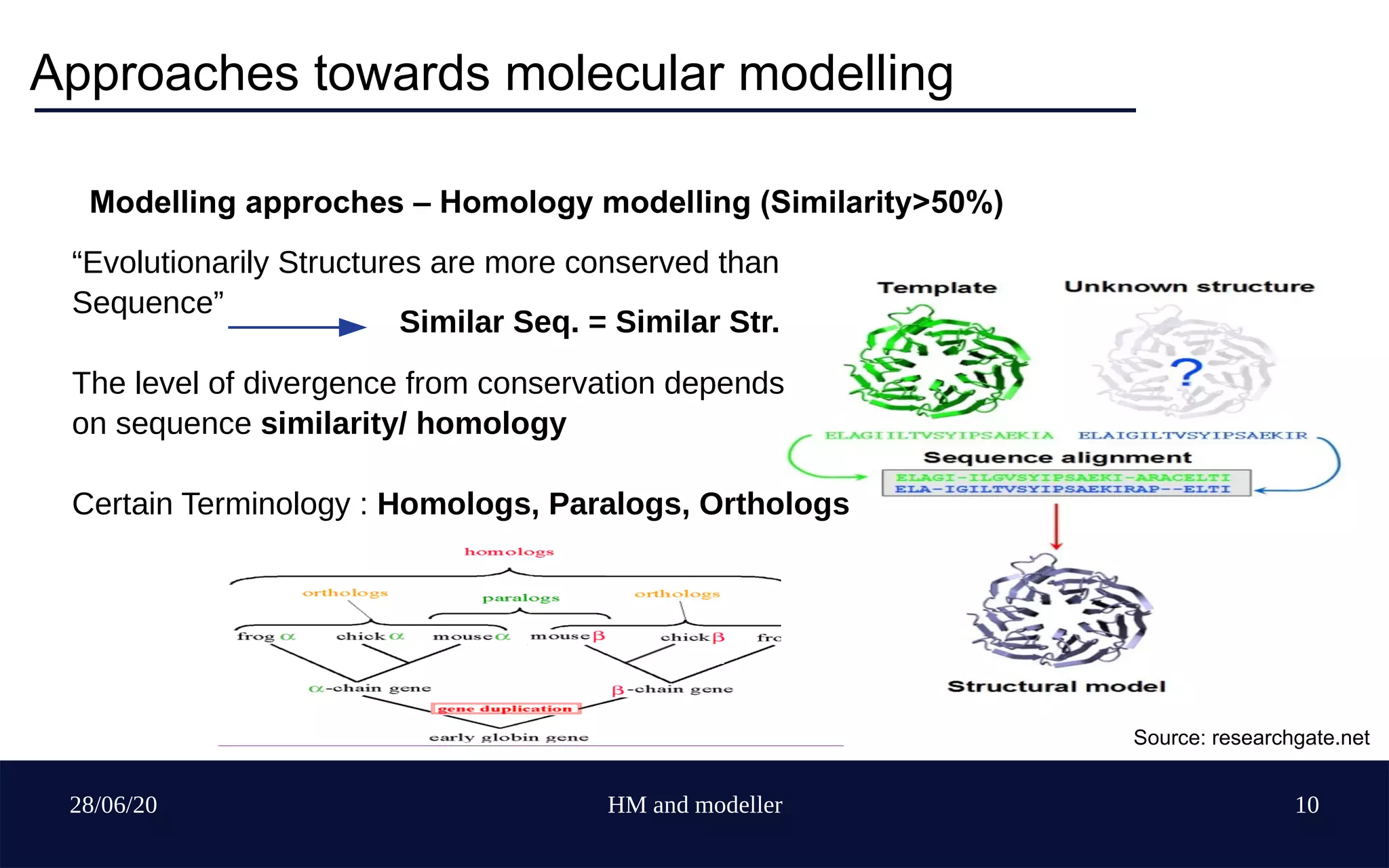
The document provides an overview of homology modeling and the use of Modeller, a protein structure prediction tool. It covers fundamental concepts of molecular modeling, various modeling approaches, limitations of protein structure acquisition, and detailed steps for using Modeller software, including installation, template selection, sequence alignment, and model evaluation. Additionally, it discusses methods to enhance accuracy and suggests resources for further learning.






![28/06/20 HM and modeller 14
Homology modelling Workflow
Read the PDB paper, Checkout the quality parameter
{Resolution,R-value, R-free Value}, Read the str.pdb
check for remarks, wwPDBValidation report
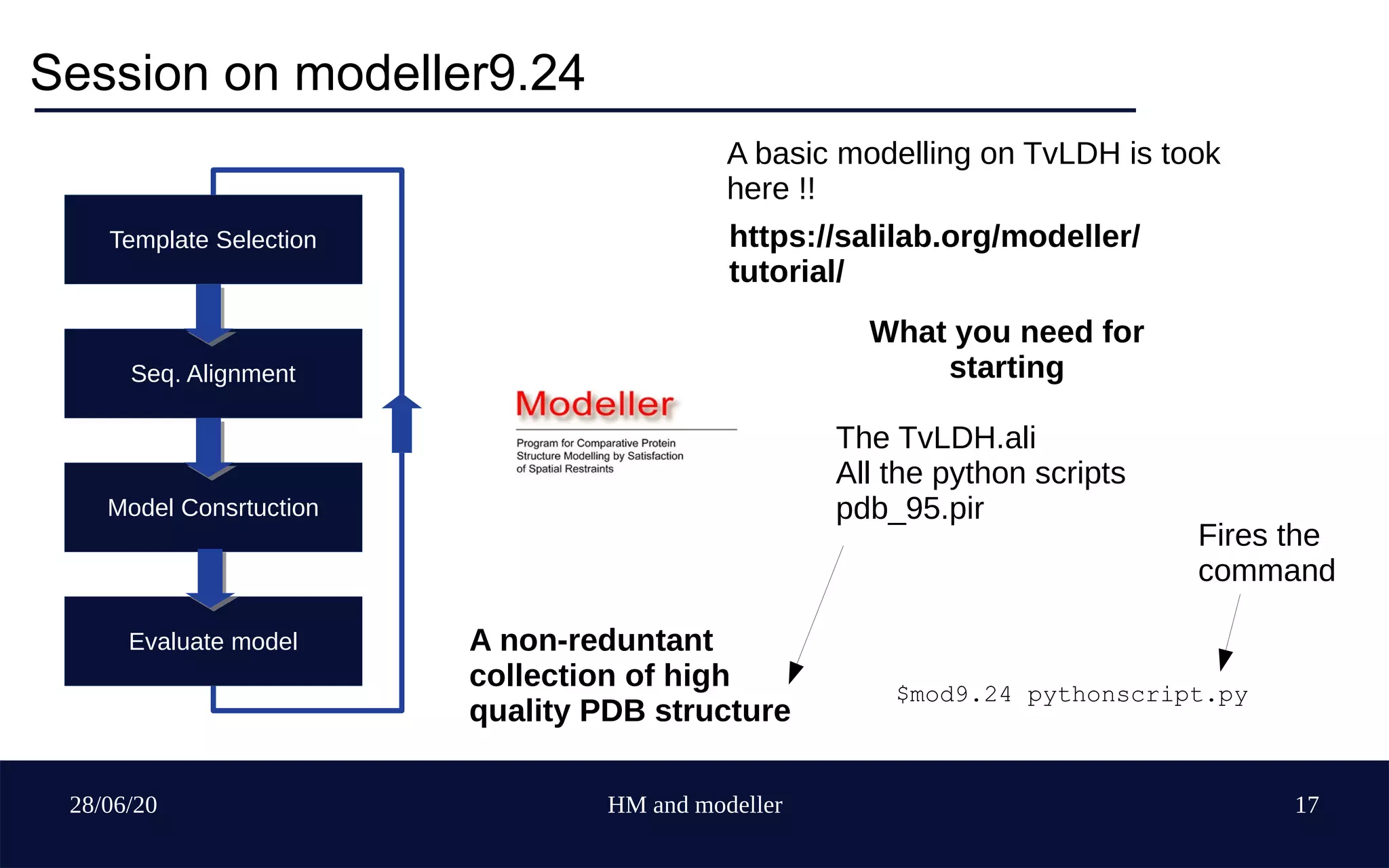
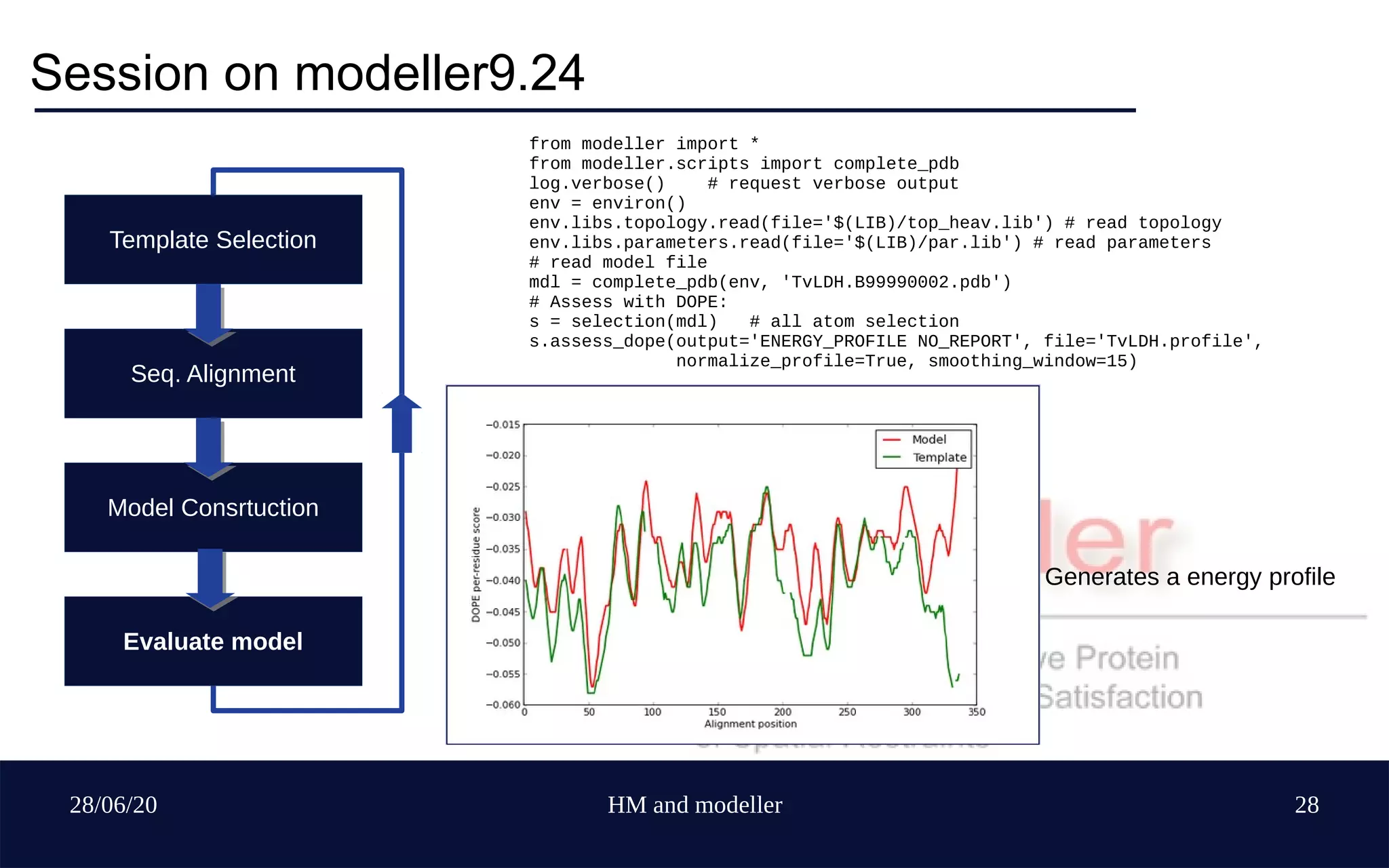
Template Selection
Seq. Alignment
Model Construction
Evaluate model
Template idenfication E-value [close to 0] > Coverage > Identity > Similarty
Srinivas Ramachandran, Pradeep Kota, Feng Ding,,Nikolay V. Dokholyan et alProteins. 2011 Jan; 79(1): 261–270.
https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=BlastHelp#get_subsequence
Davis, A.M., Teague, S.J. and Kleywegt, G.J. (2003), Angewandte Chemie International Edition, 42: 2718-2736.
https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/r-value-and-r-free
Sequence based and Structure based
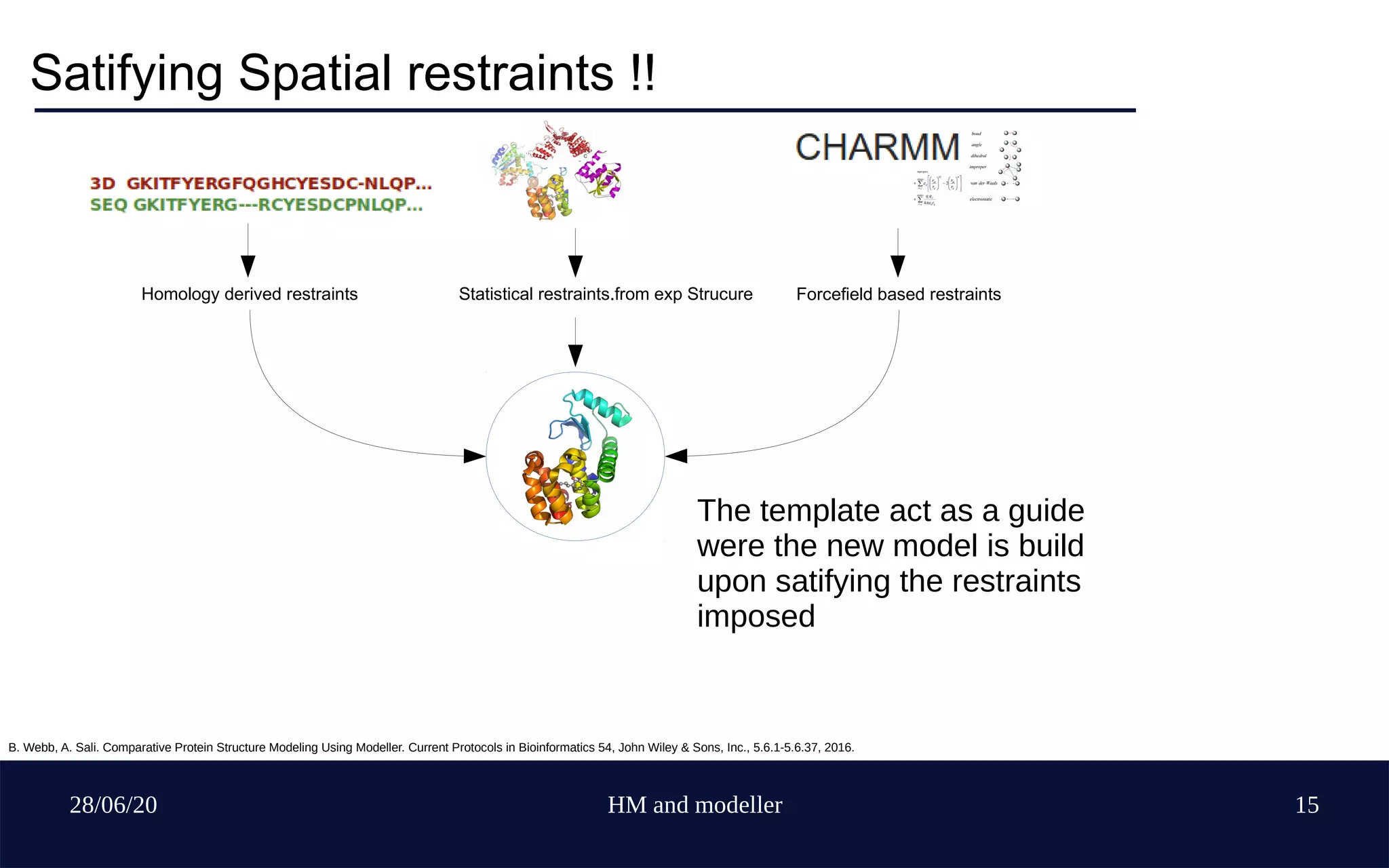
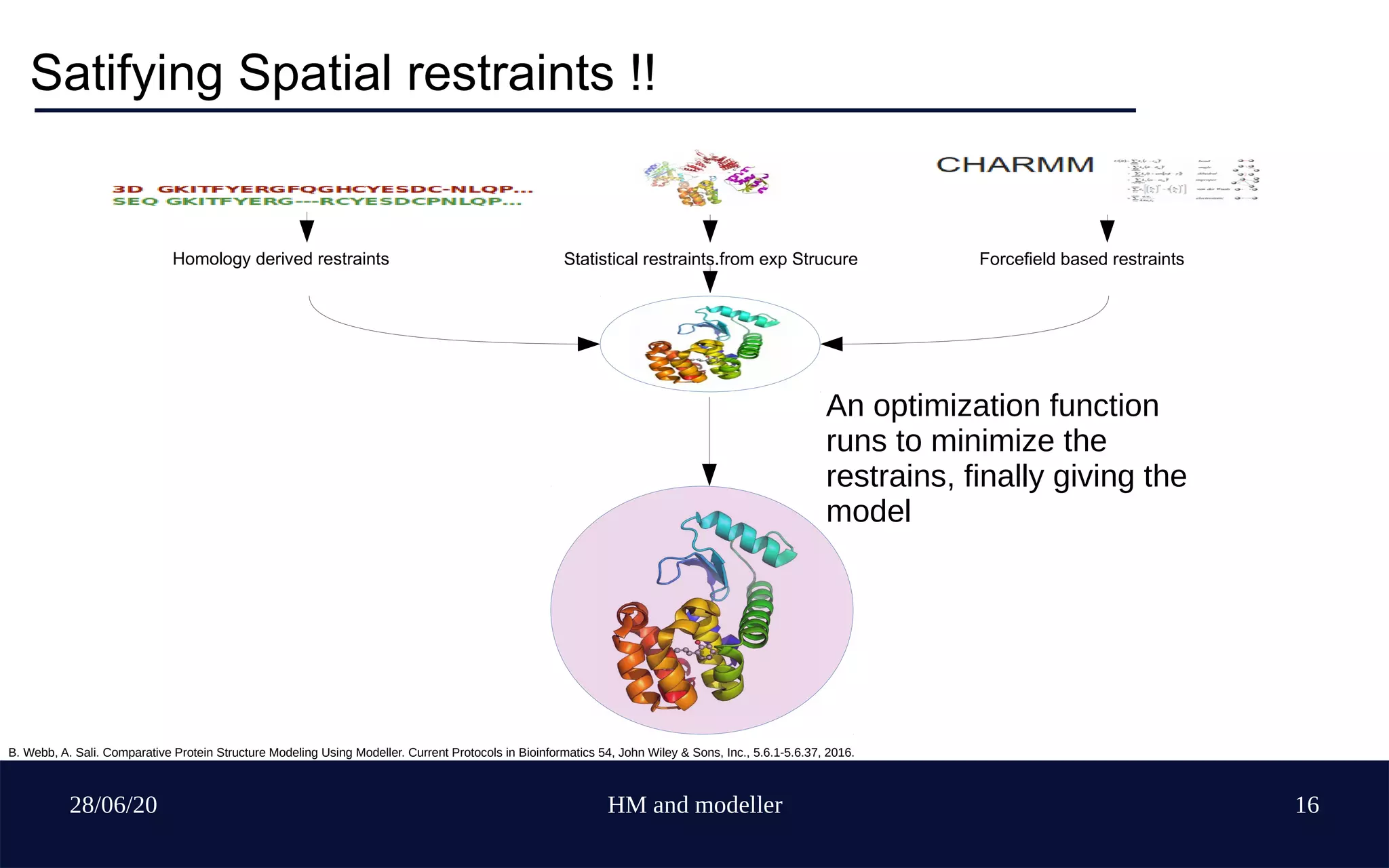
Rigid body assembly, segment matching and satisfacti
-on of spatial restraints method !!
Swiss model validation, SAVES, PROSESS](https://image.slidesharecdn.com/homologymodeling-200628023333/75/Homology-modeling-14-2048.jpg)