Download as PDF, PPTX






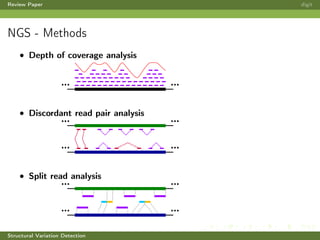

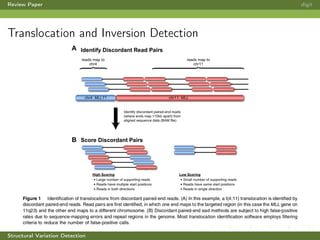

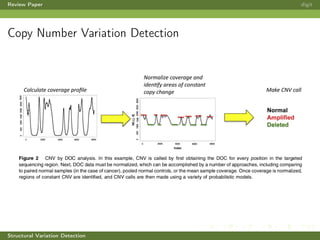








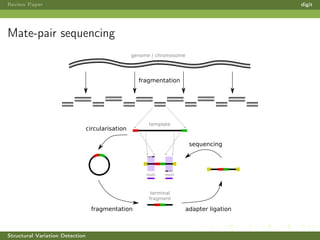
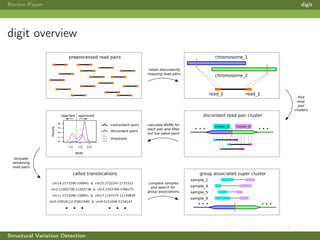

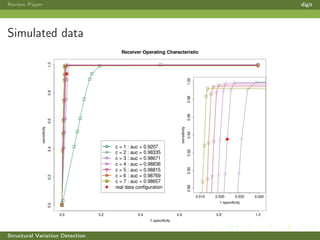
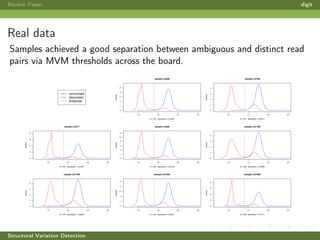


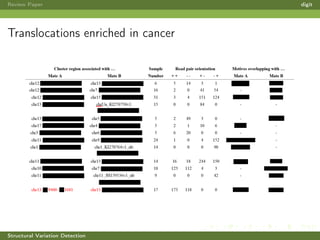



This review paper focuses on the detection of structural DNA variations using various informatic approaches, highlighting techniques such as next-generation sequencing (NGS), cytogenetics, and microarrays. It discusses the strengths and weaknesses of different methods for detecting translocations, inversions, copy number variations, and insertions/deletions, emphasizing the need for complementary tools and extensive validation for clinical applications. The paper also presents a new tool for detecting interchromosomal translocations, demonstrating its effectiveness in reducing false positives and analyzing population-specific translocation profiles.
































































































