Download as PDF, PPTX

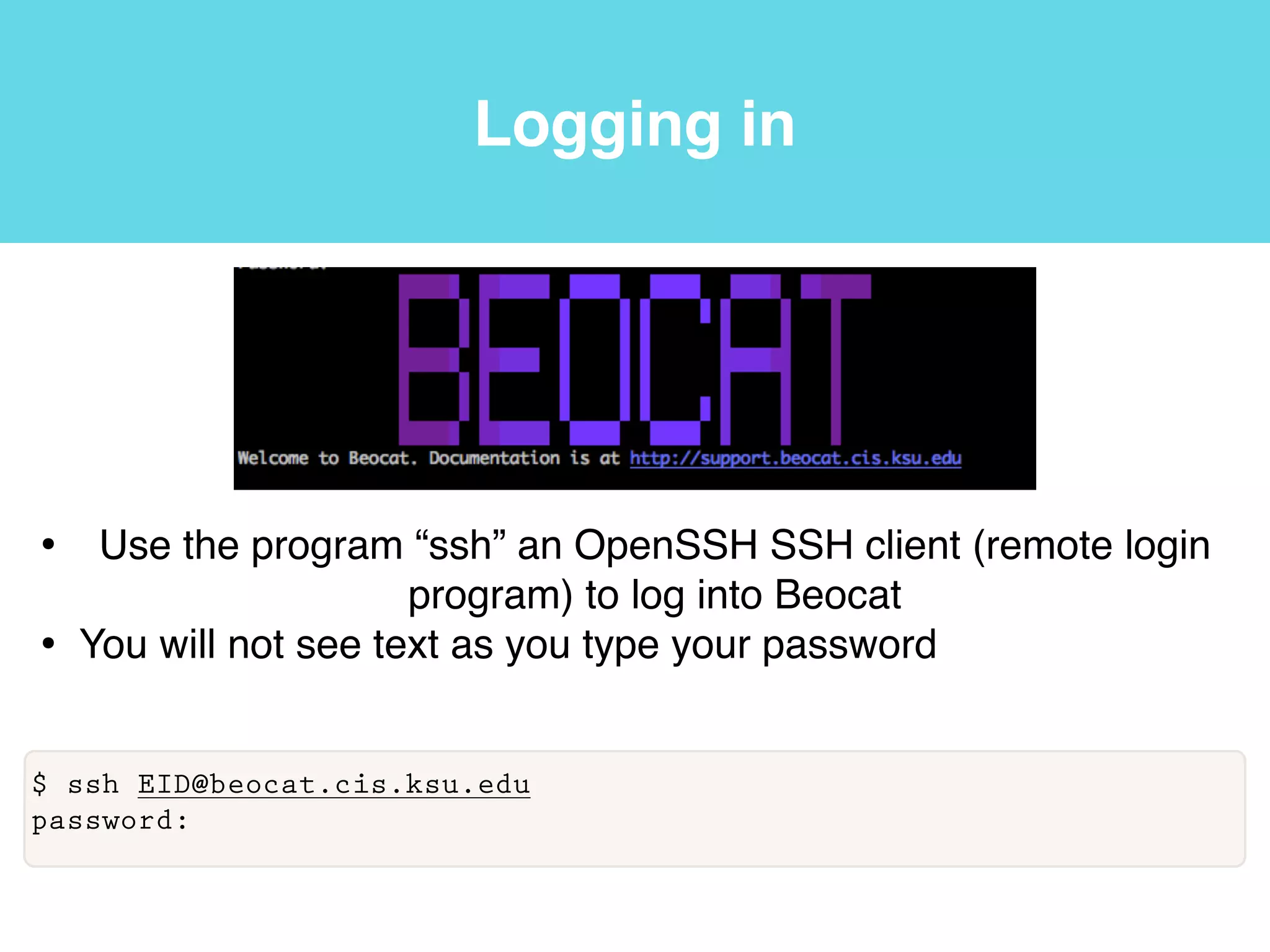

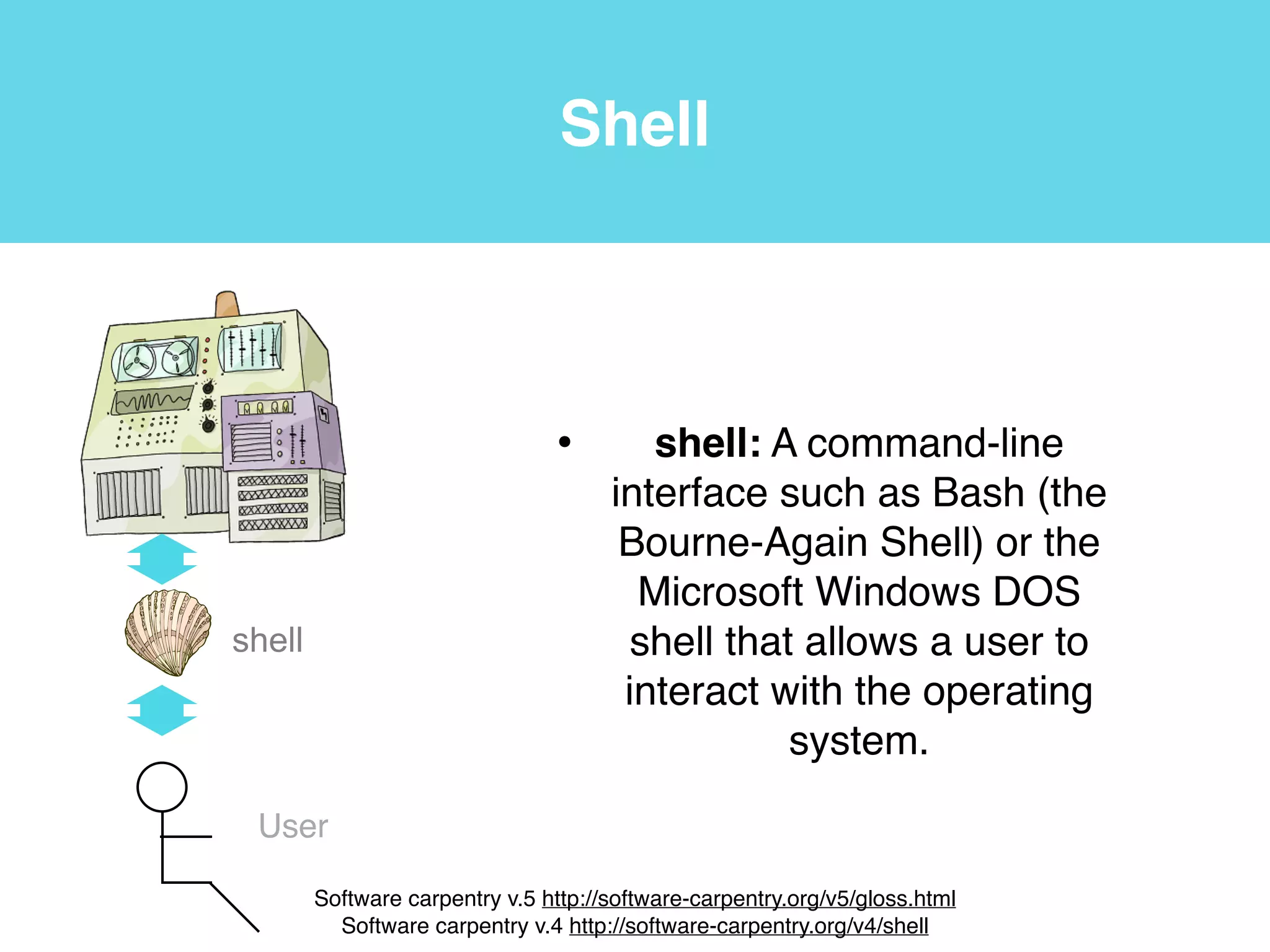
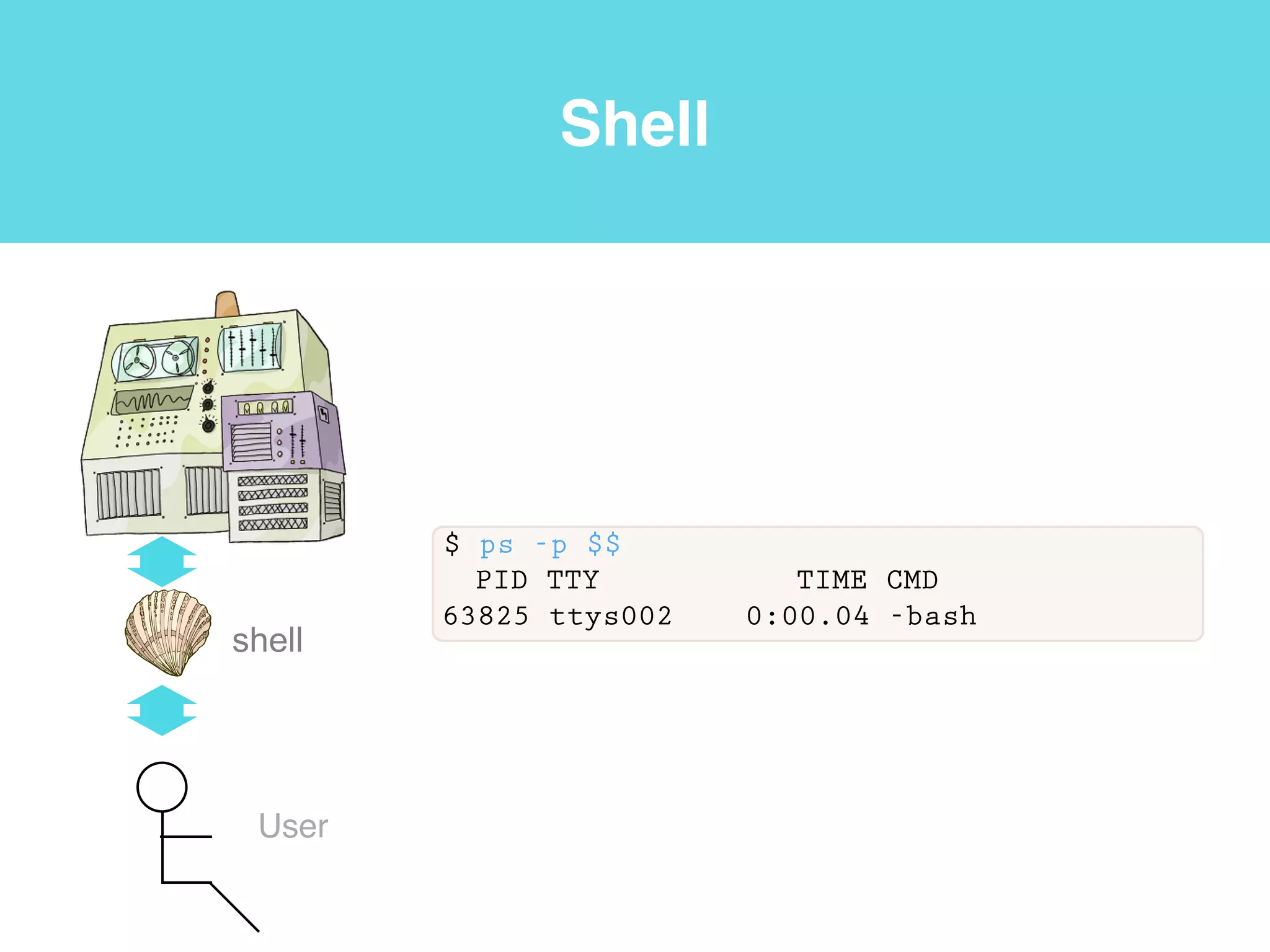
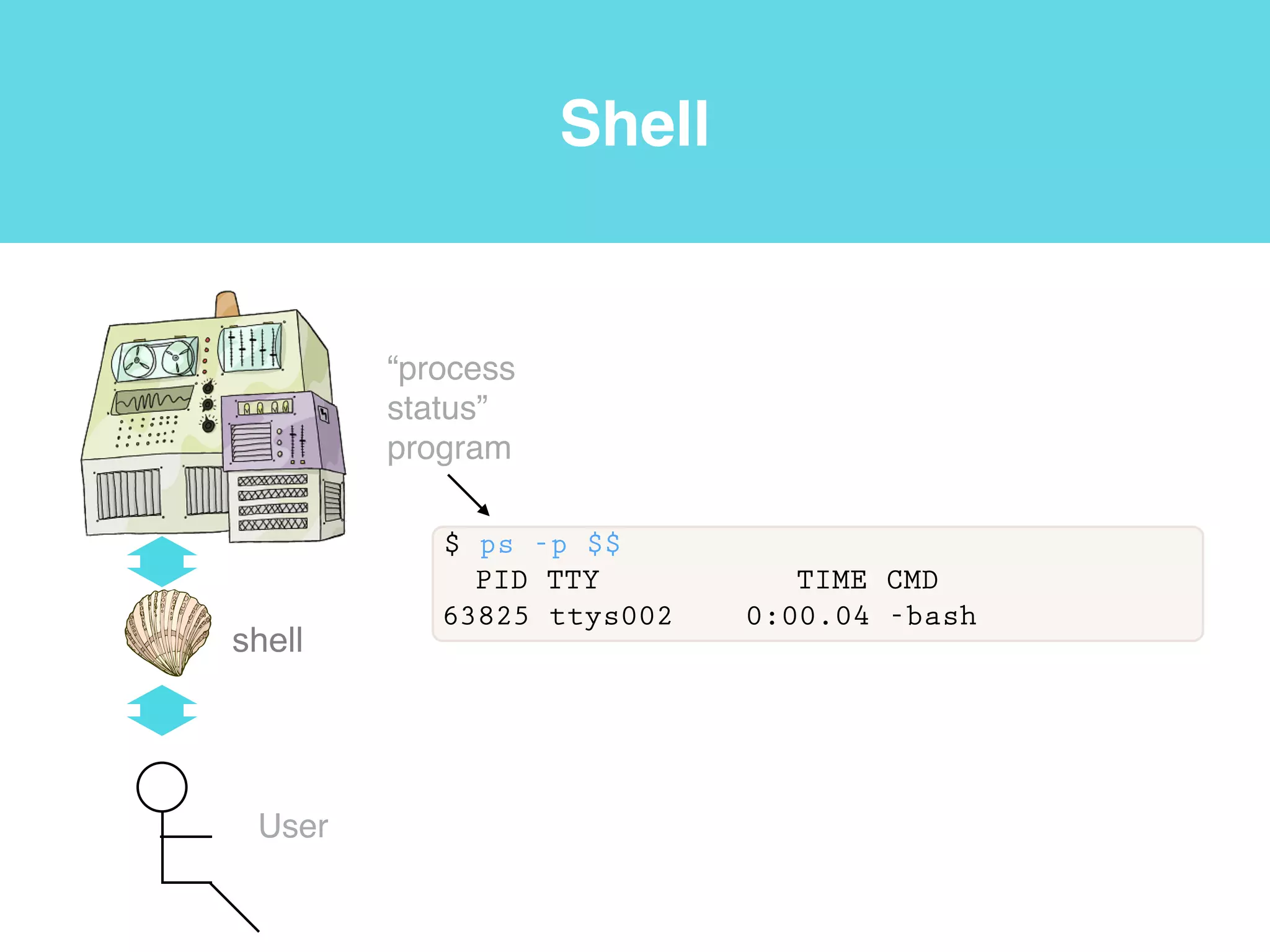
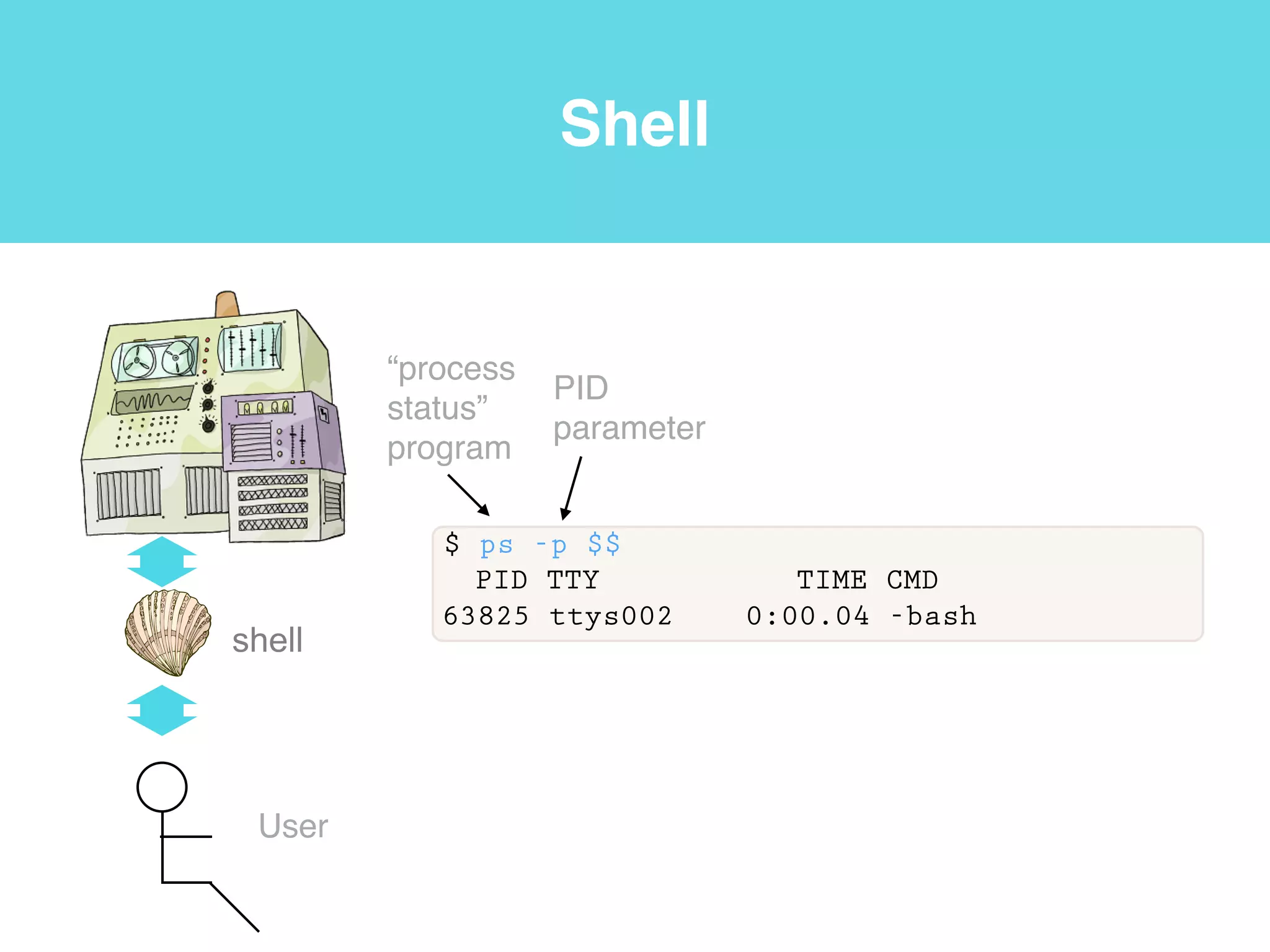
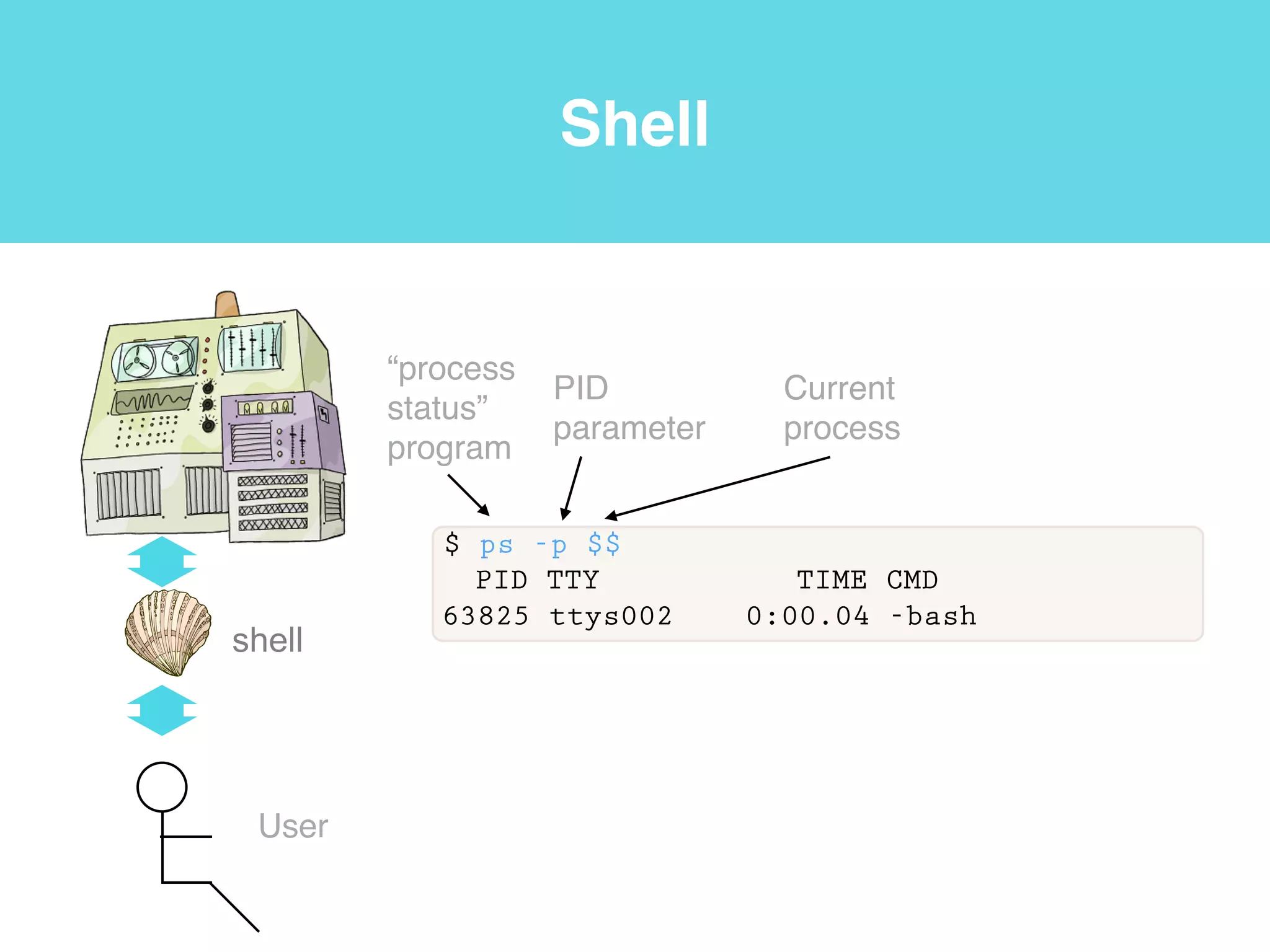
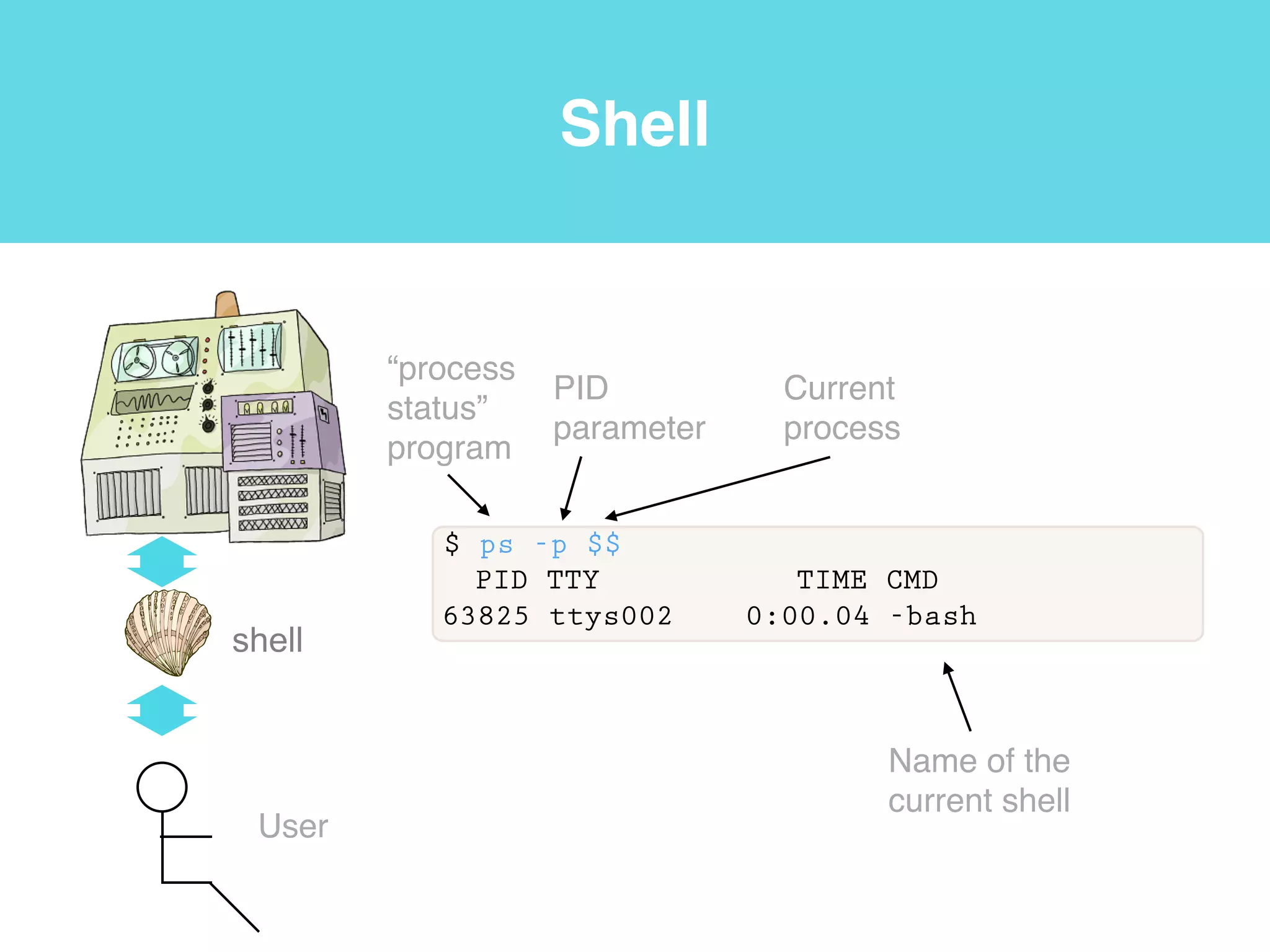
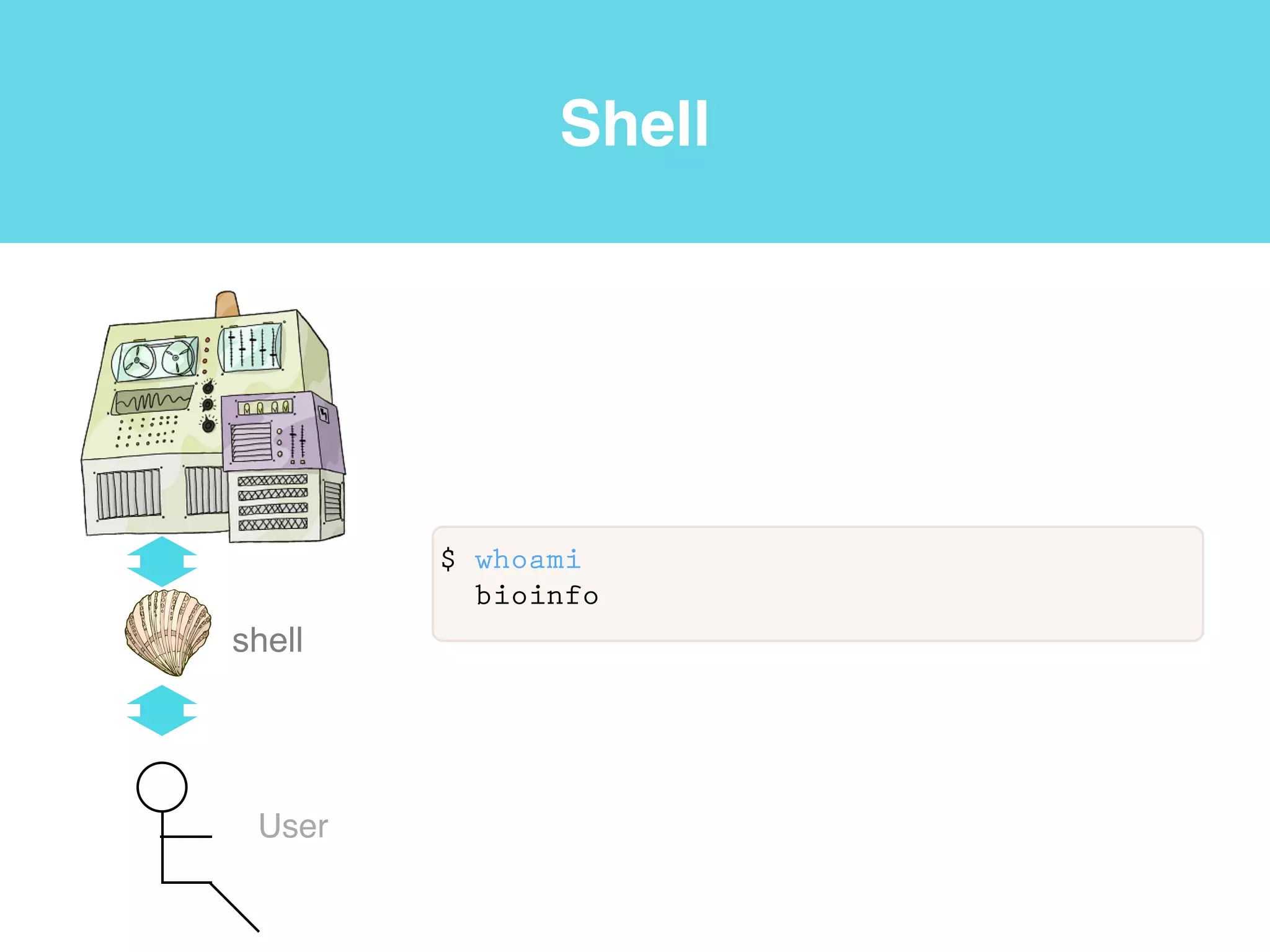
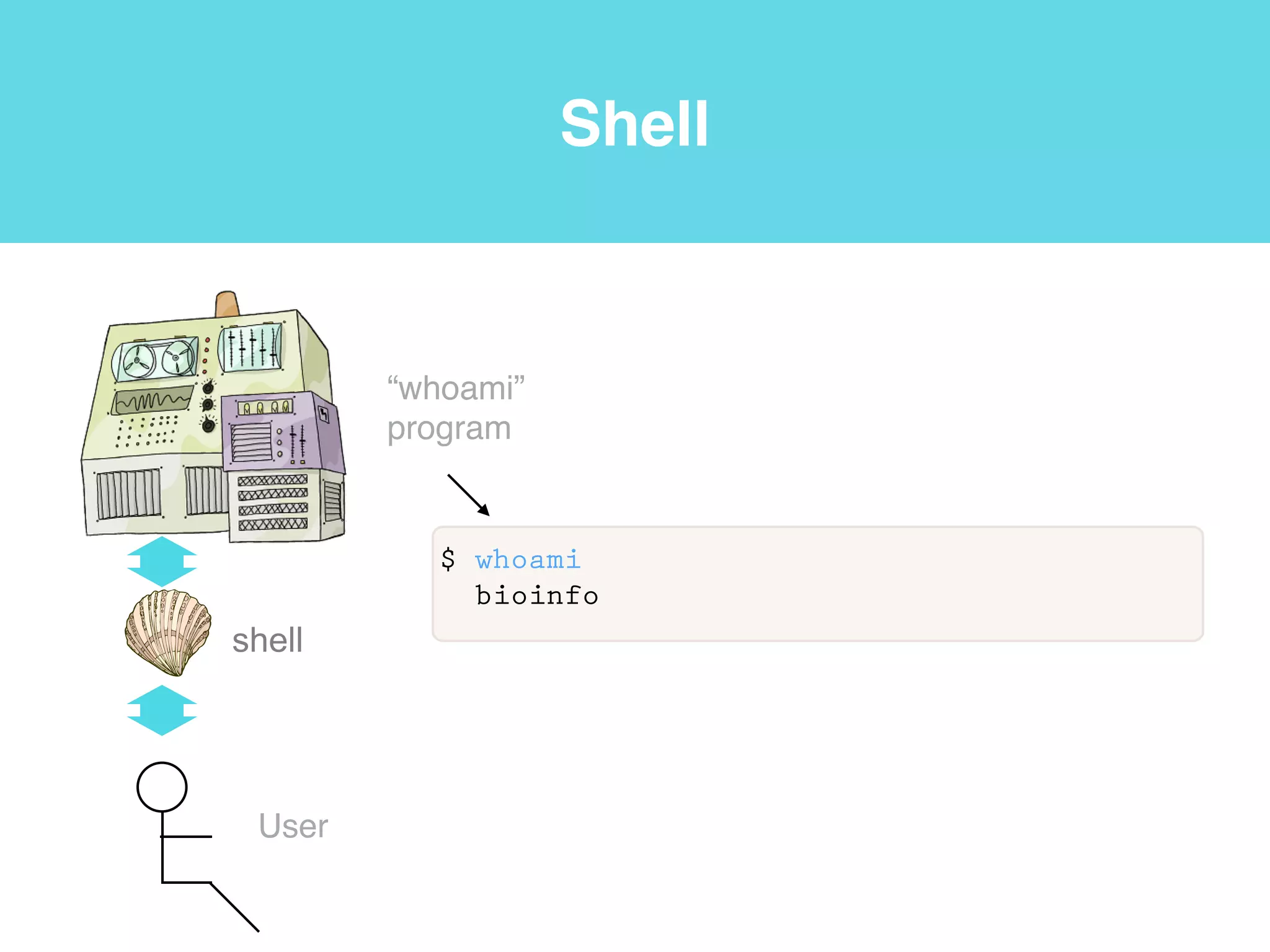
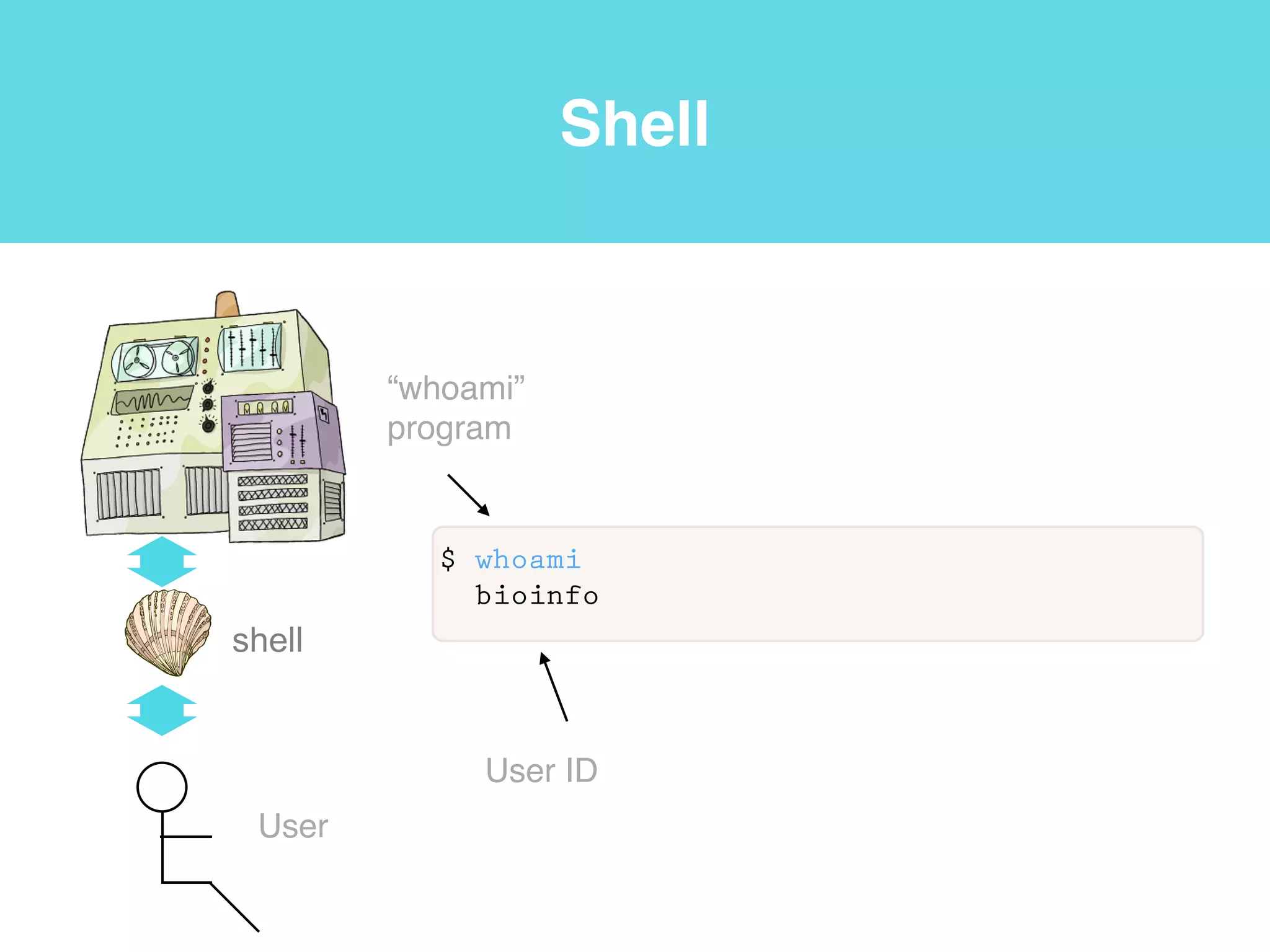
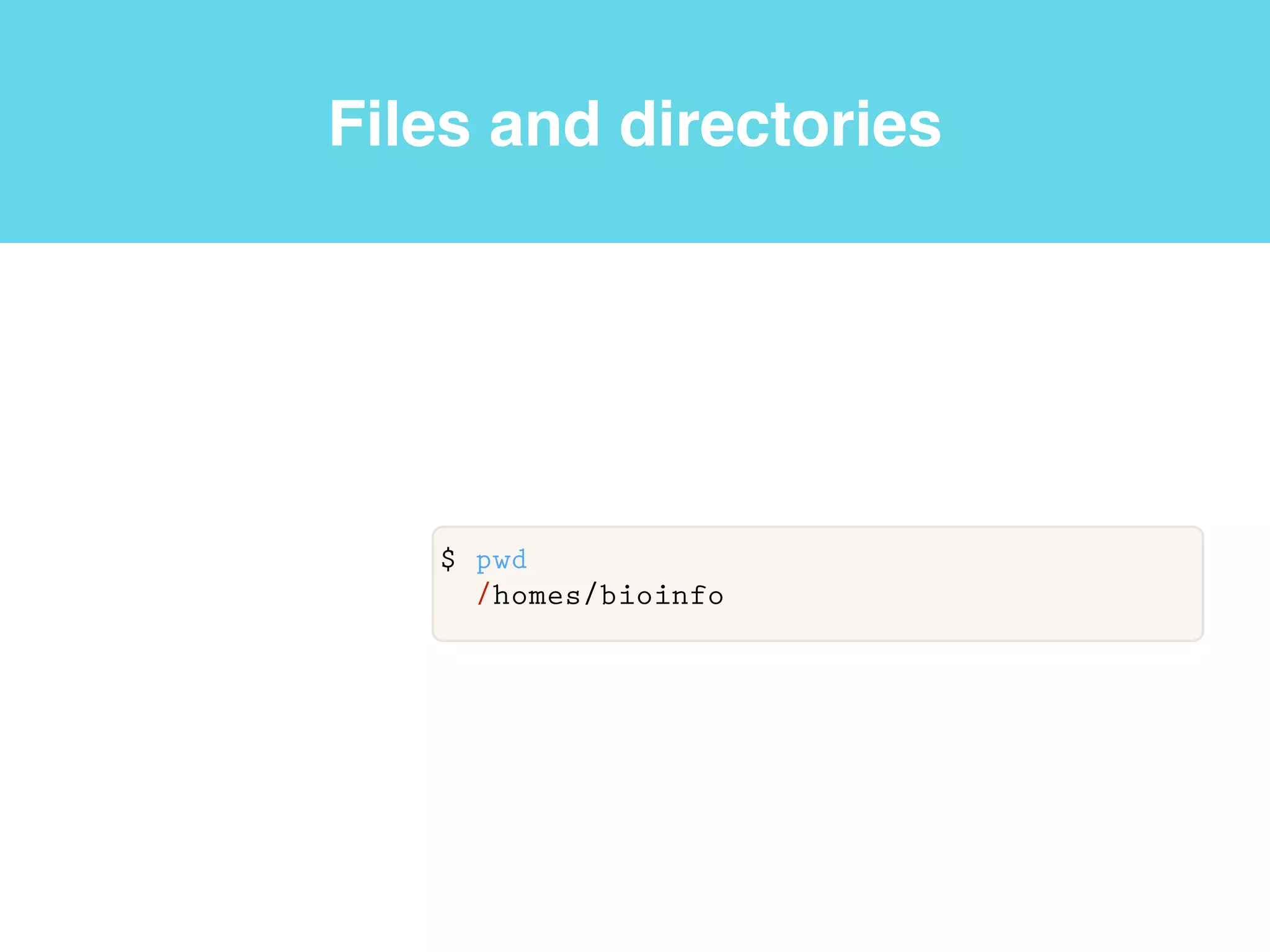



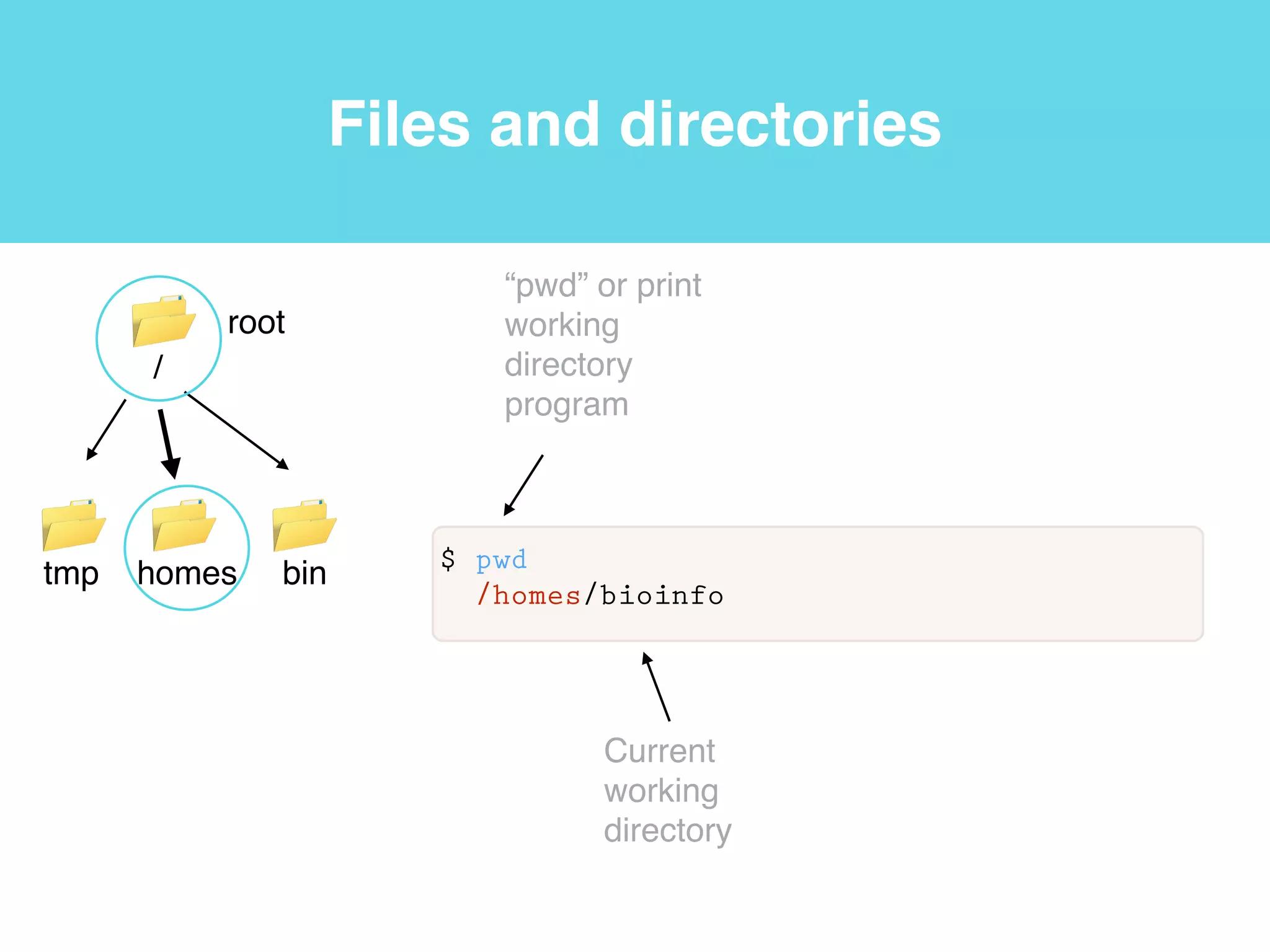
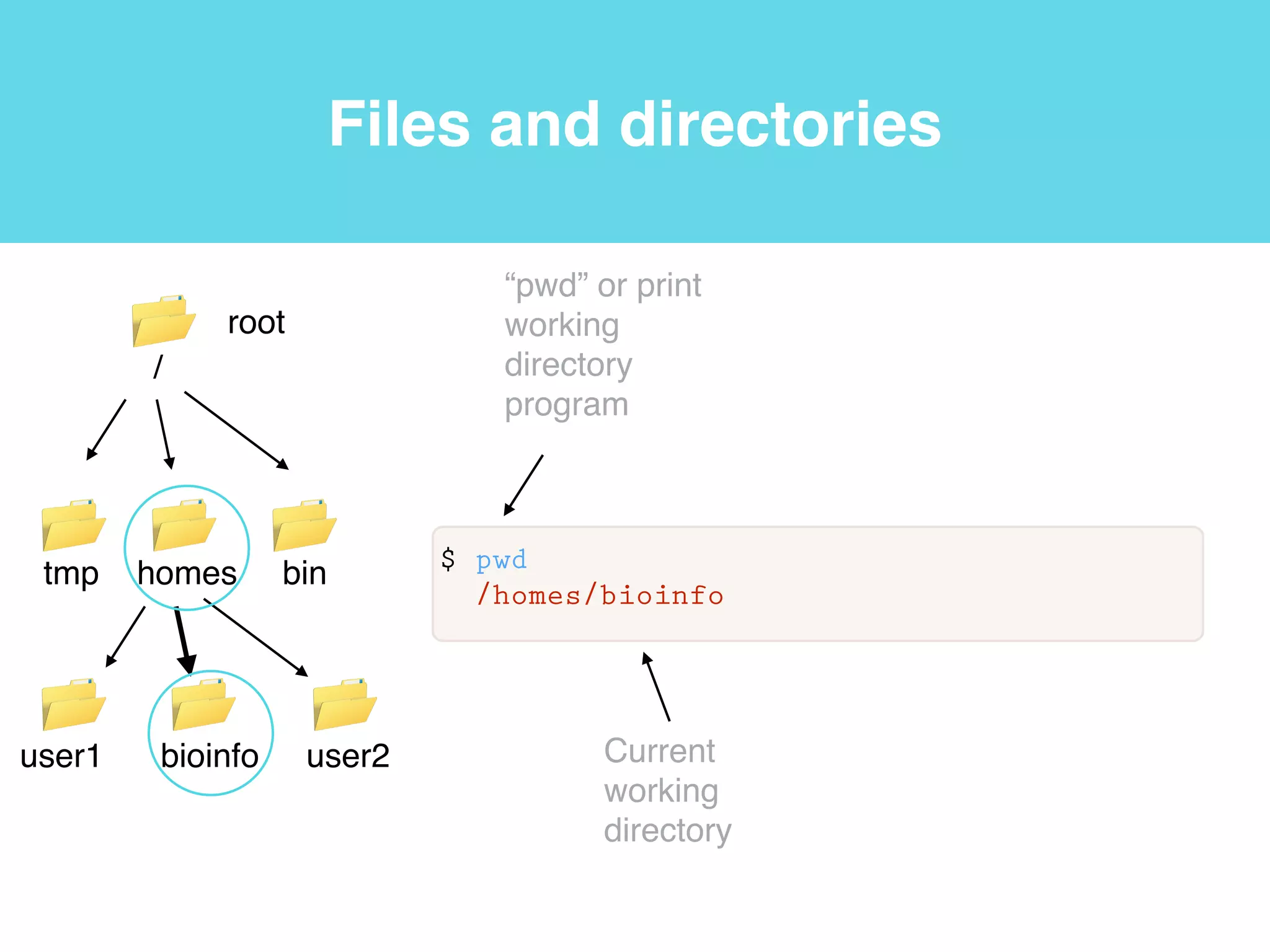




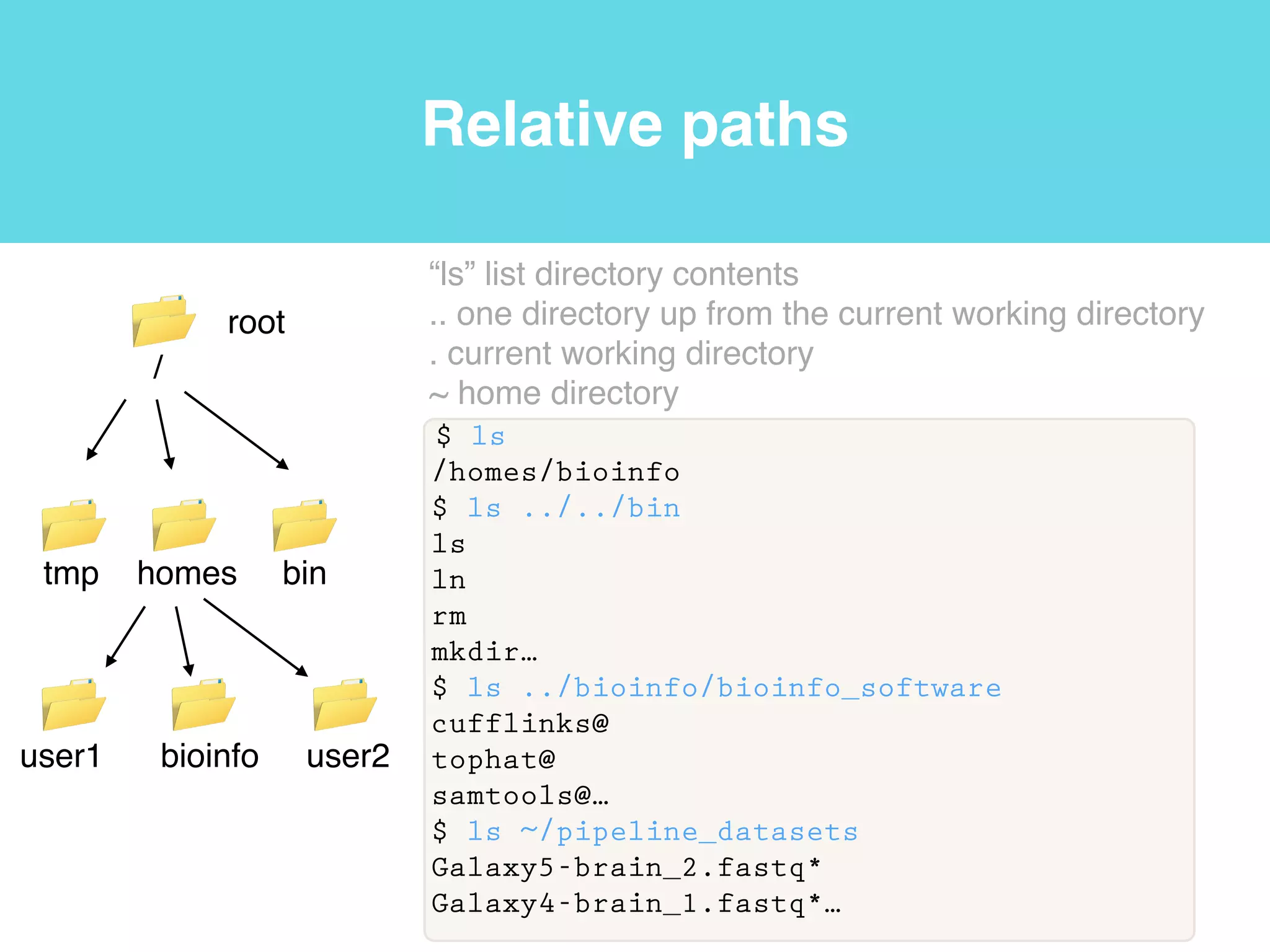
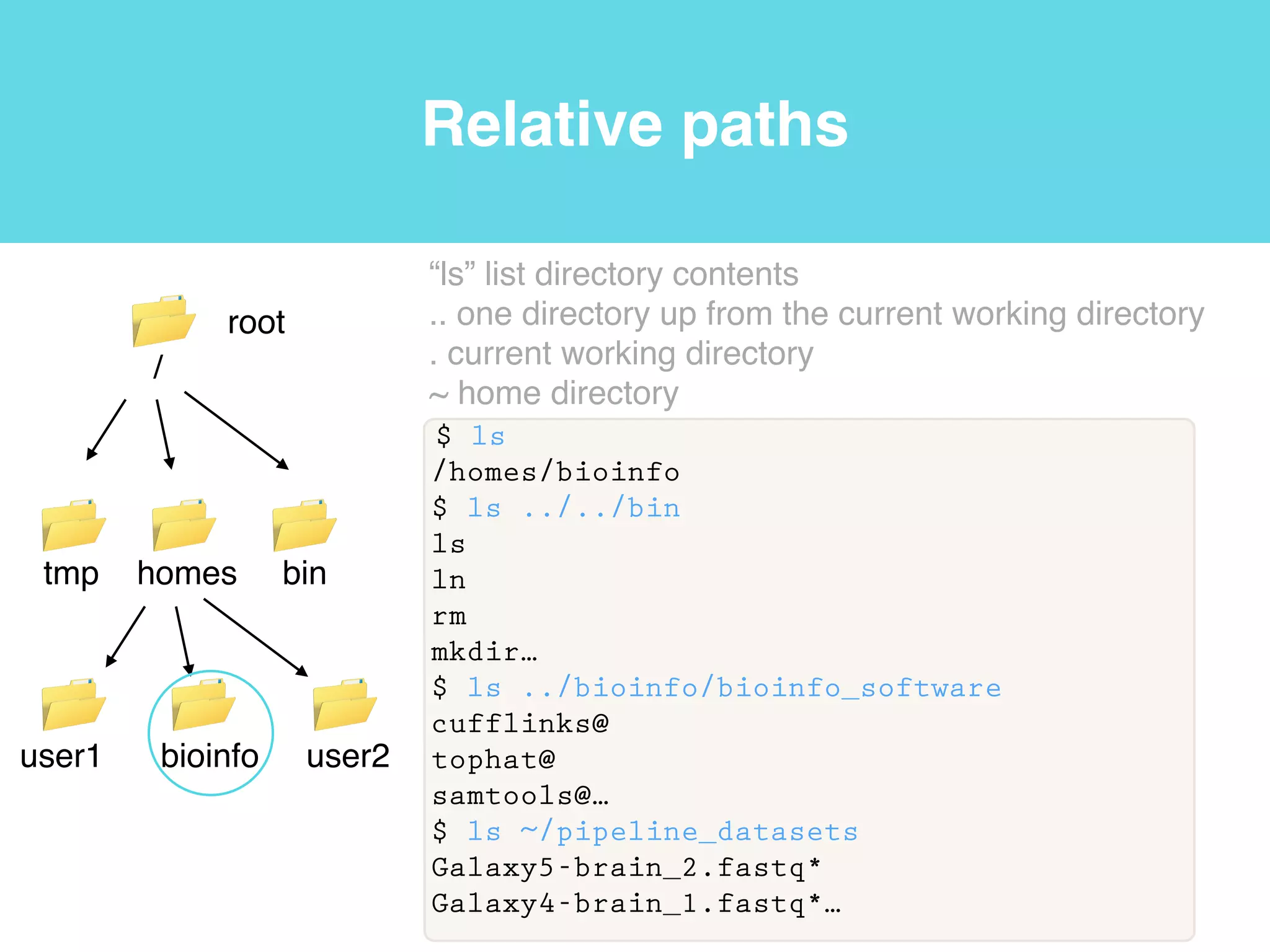
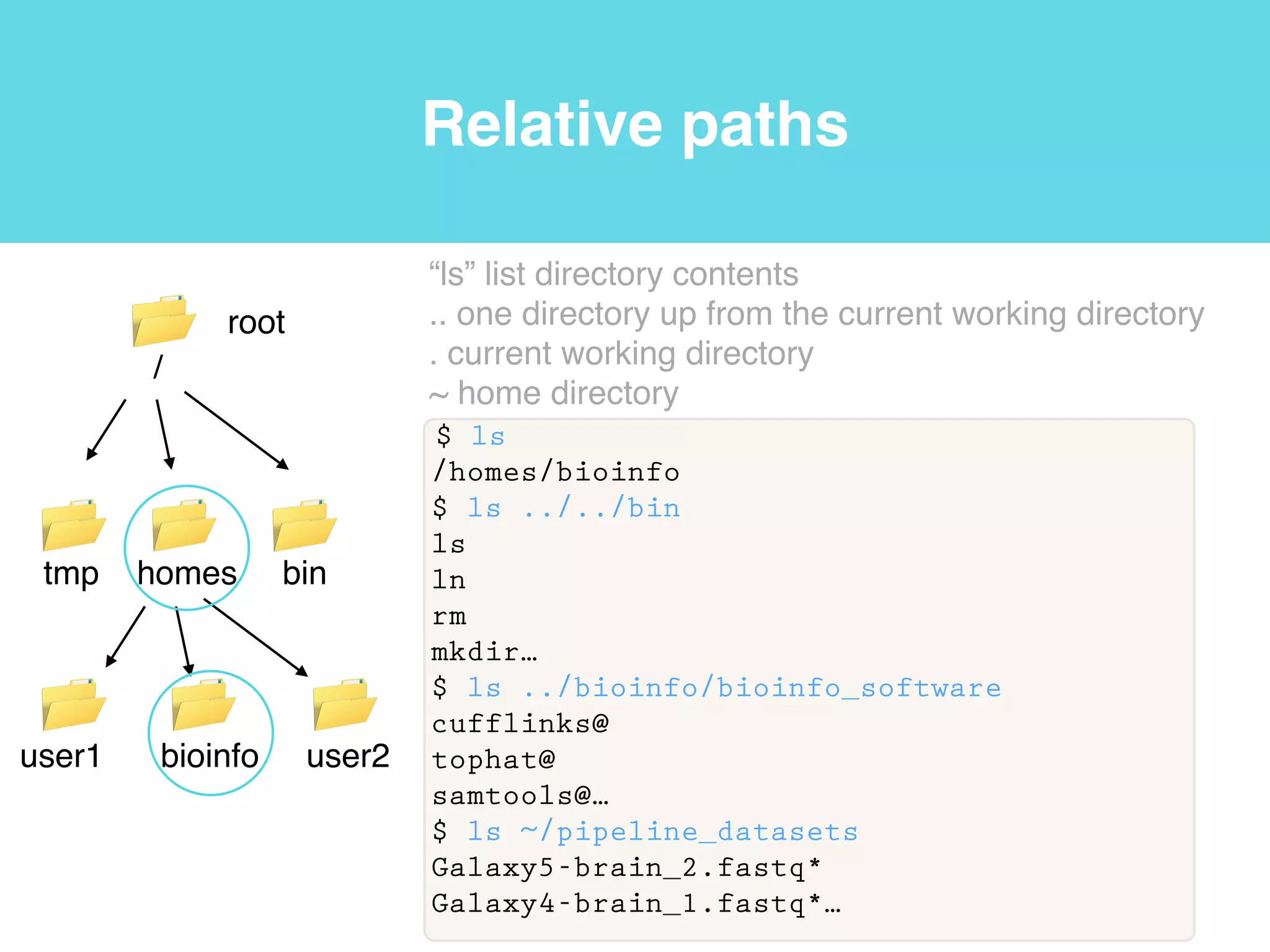
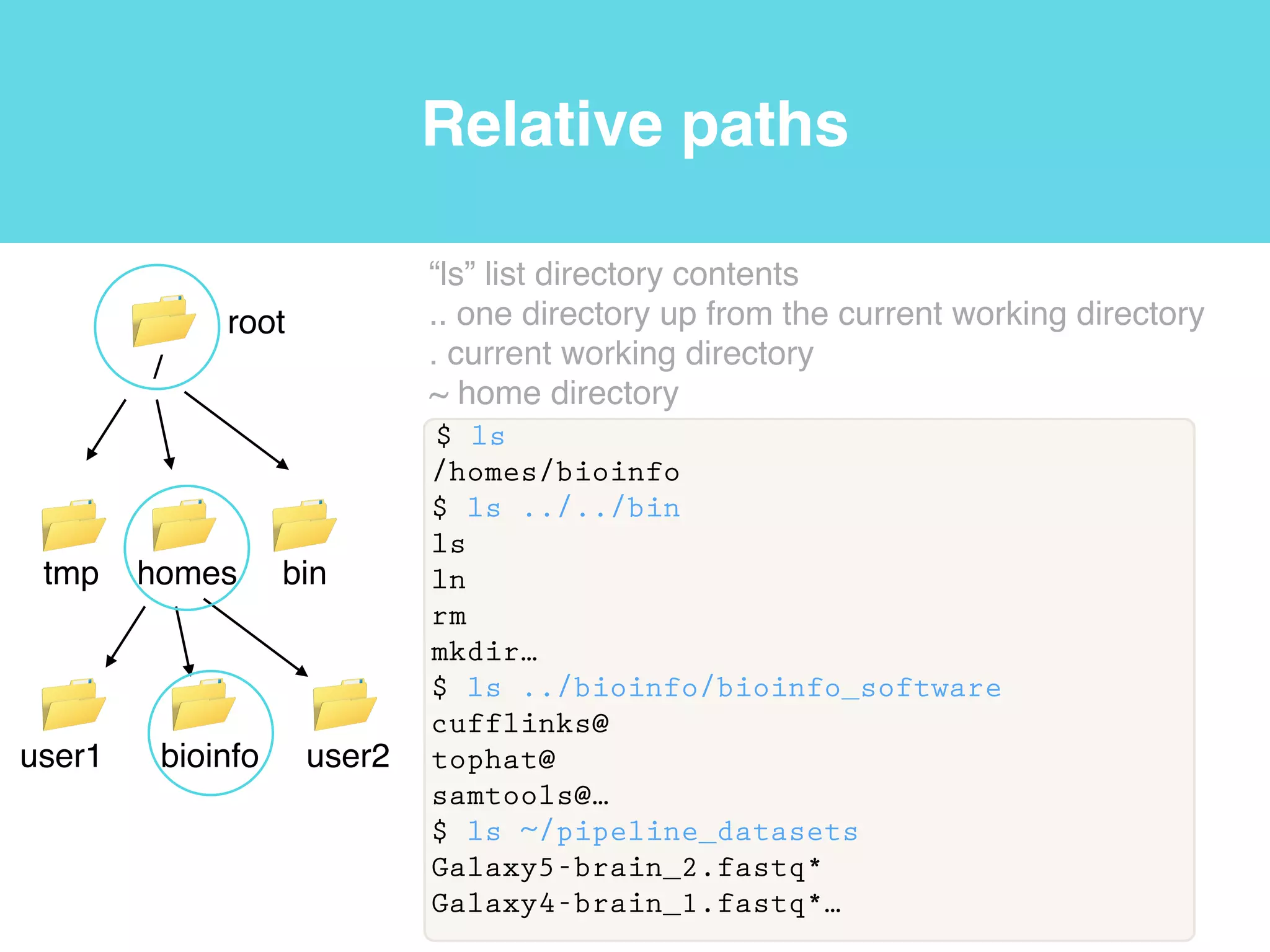
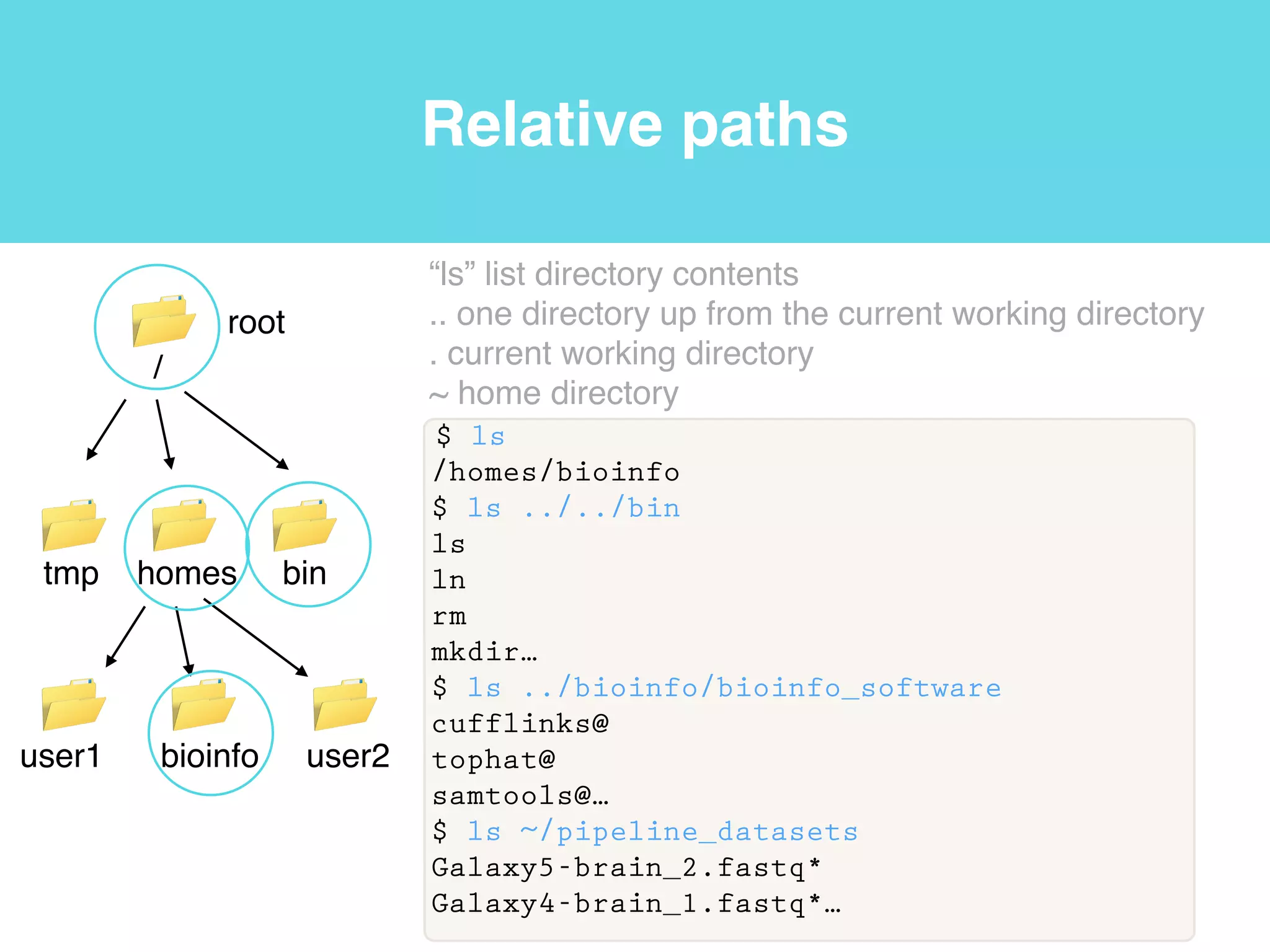
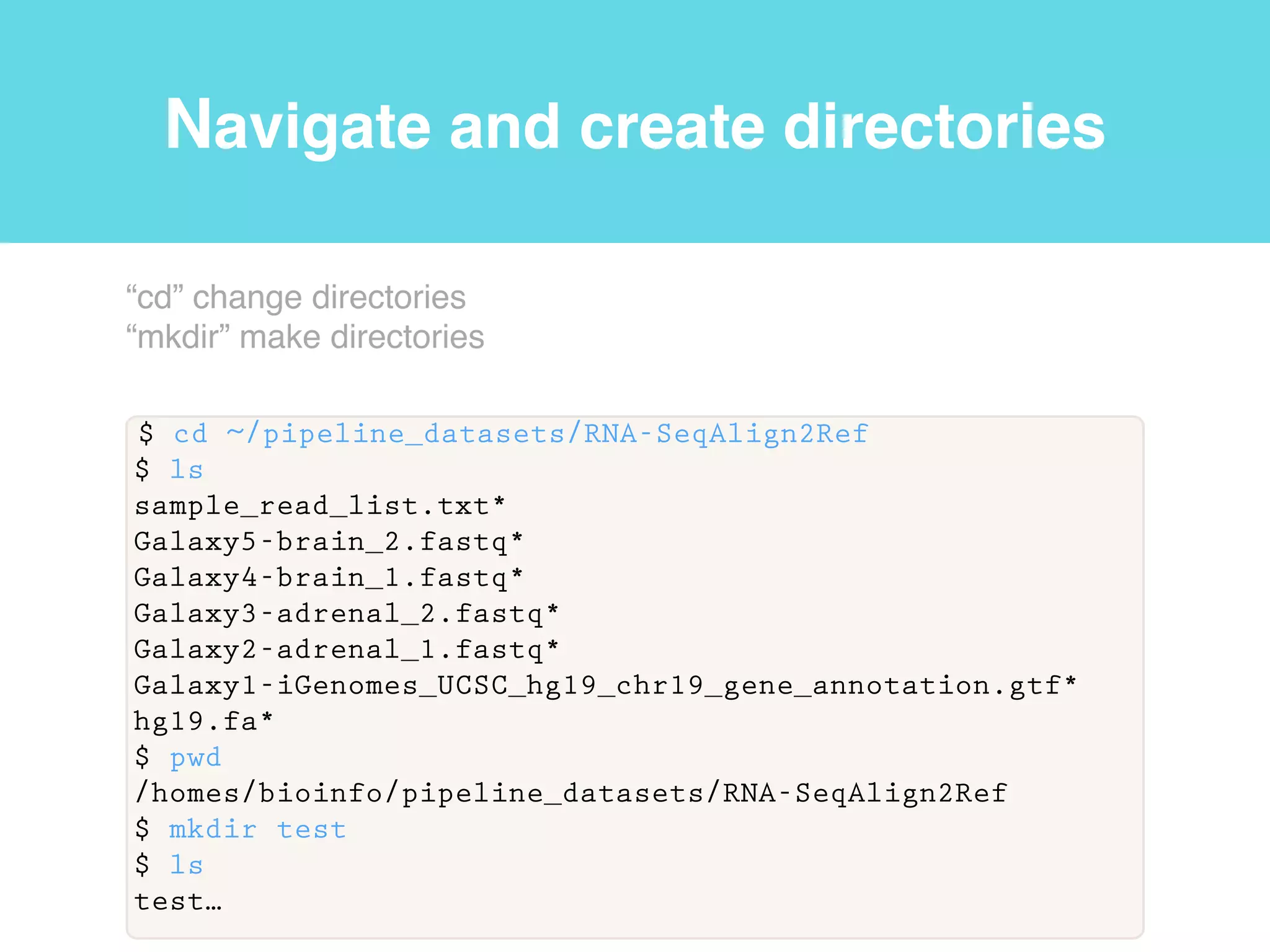
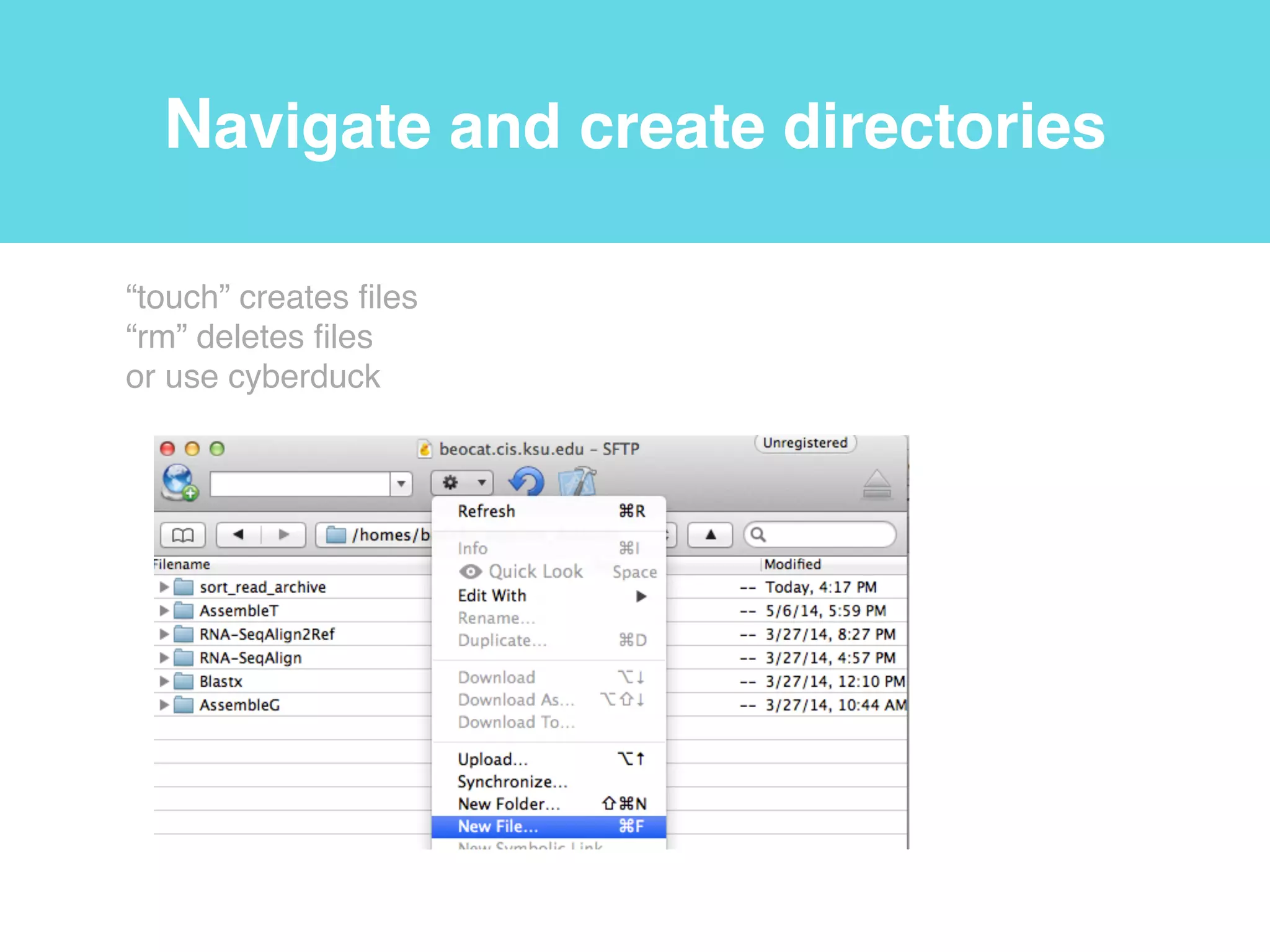
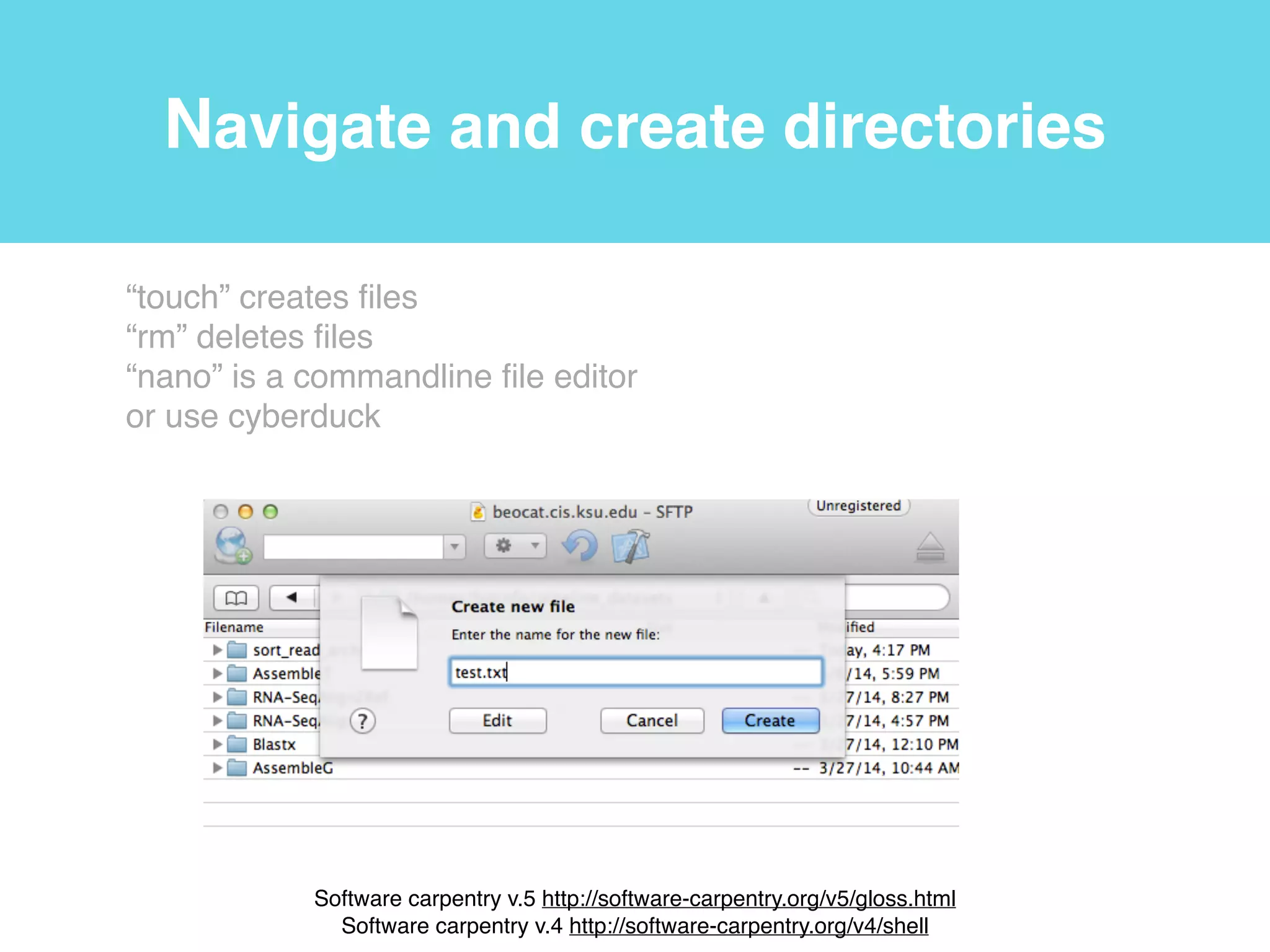
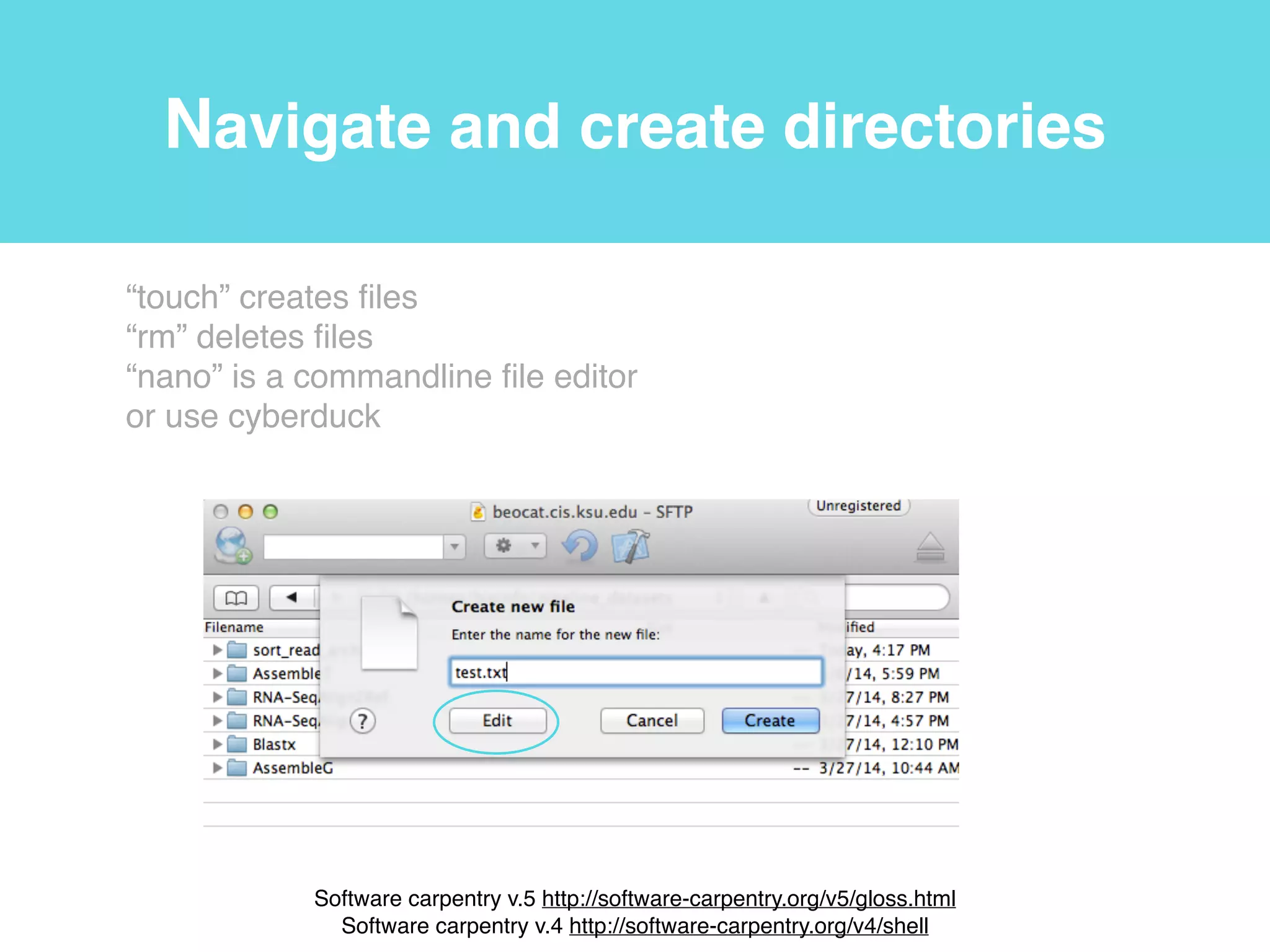
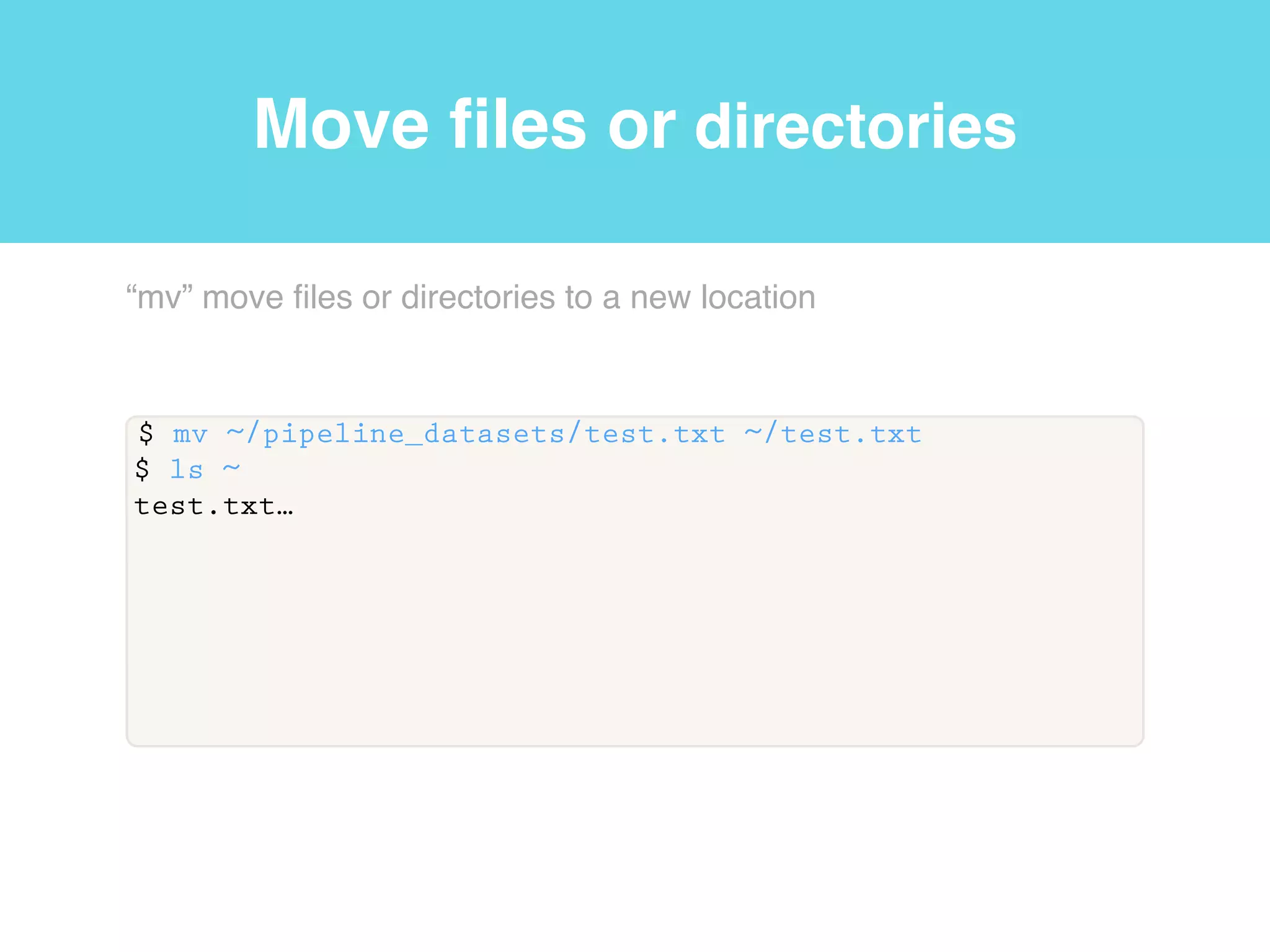


























This document provides an introduction to using the command line interface on the Beocat computing cluster. It discusses how to log in via SSH, navigate directories using commands like pwd, ls, cd, and create/delete directories and symbolic links. The document also covers using relative paths, editing files with nano, and moving files with mv.









![[COSCUP 2021] LLVM Project: The Good, The Bad, and The Ugly](https://cdn.slidesharecdn.com/ss_thumbnails/coscup2021-llvm-210731054723-thumbnail.jpg?width=640&height=640&fit=bounds)






















































































