

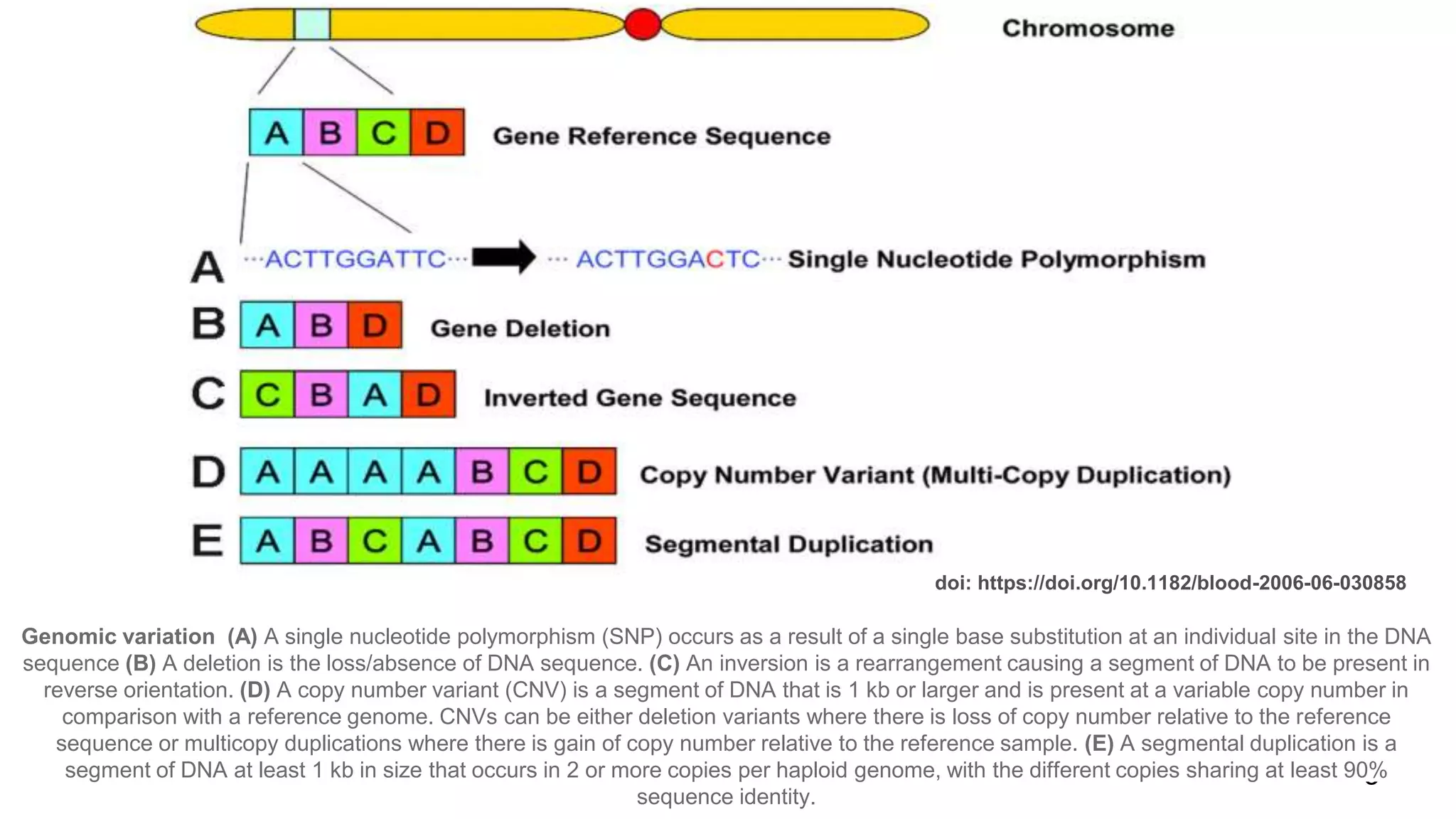






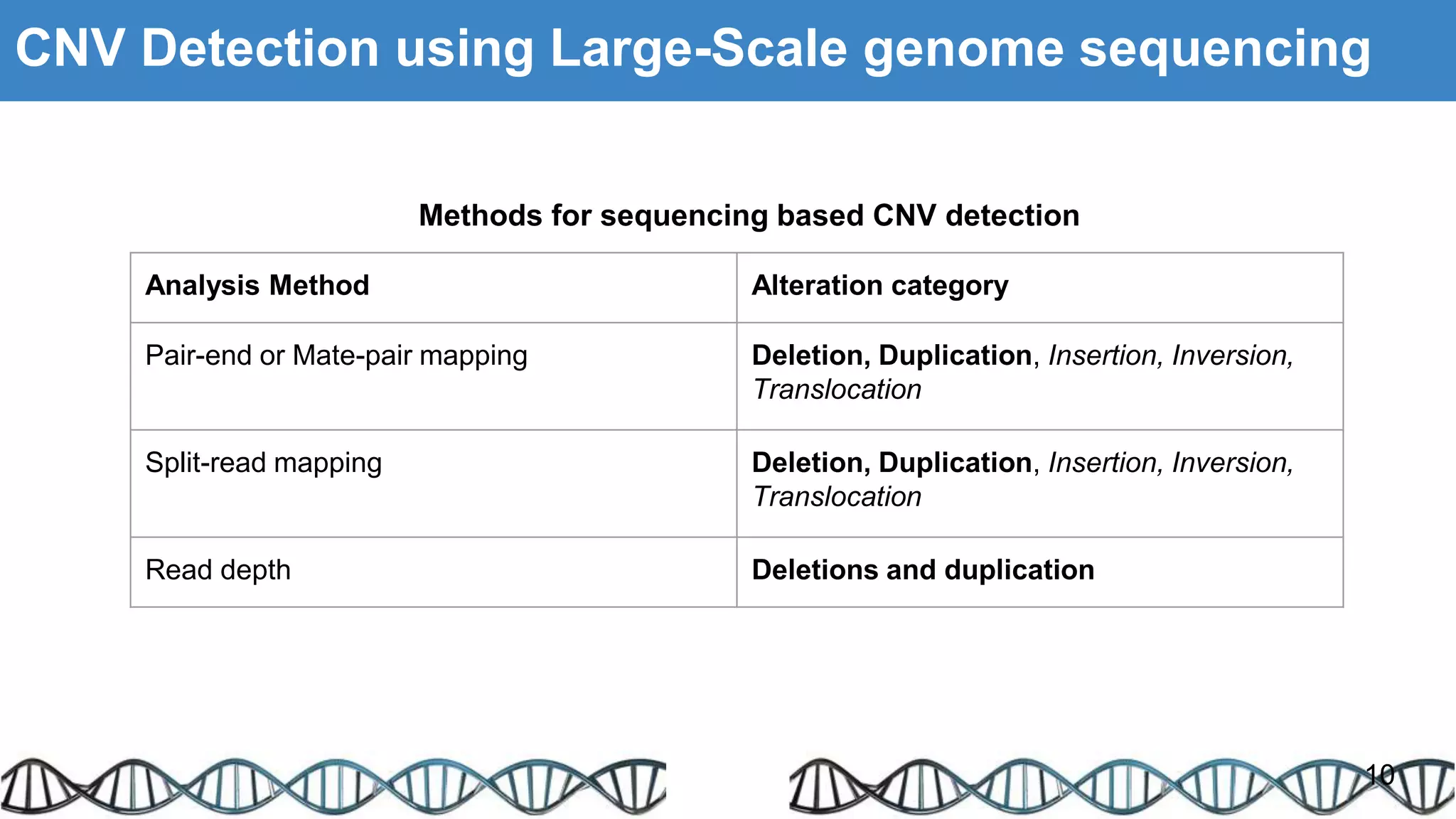
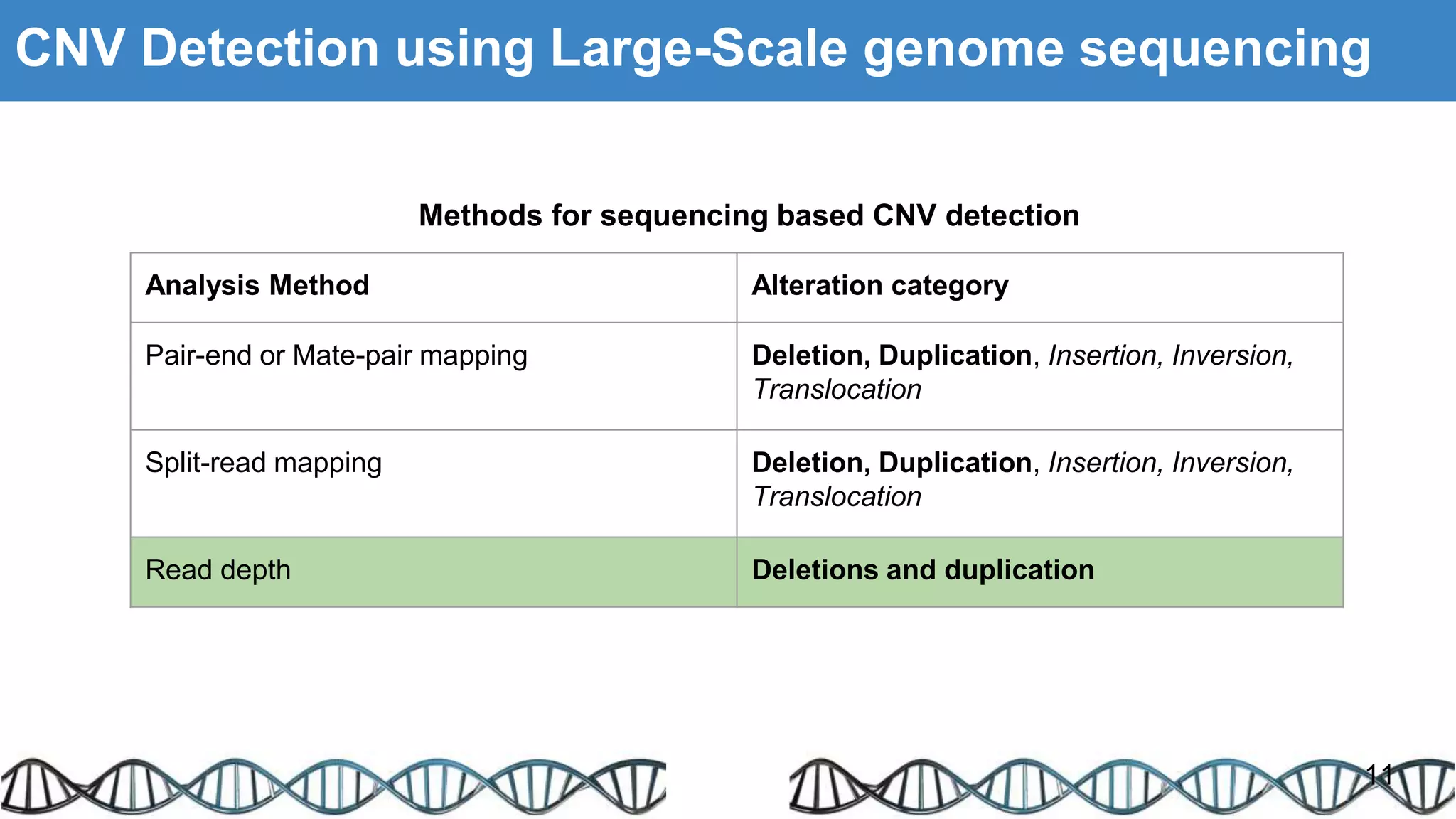
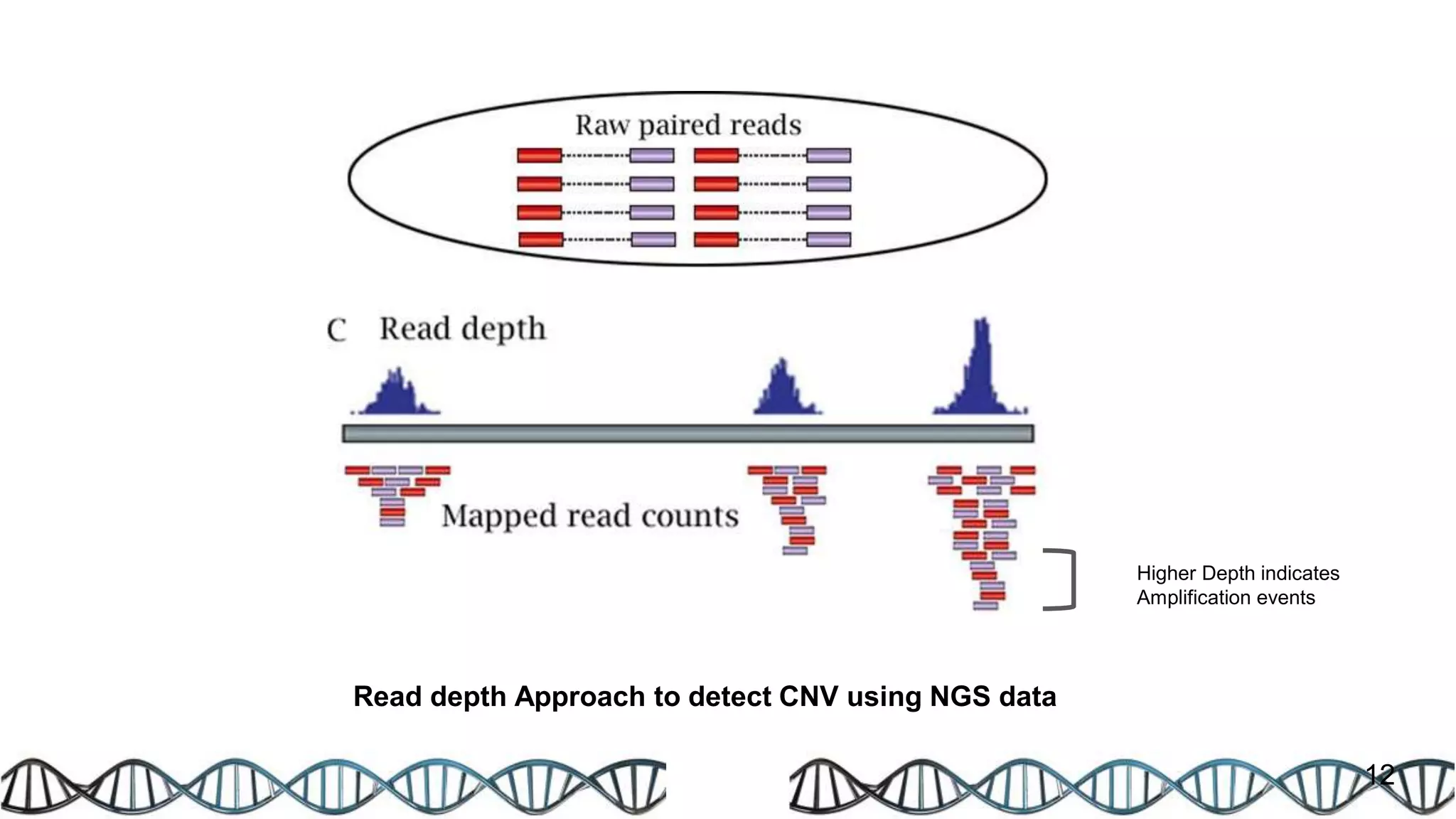
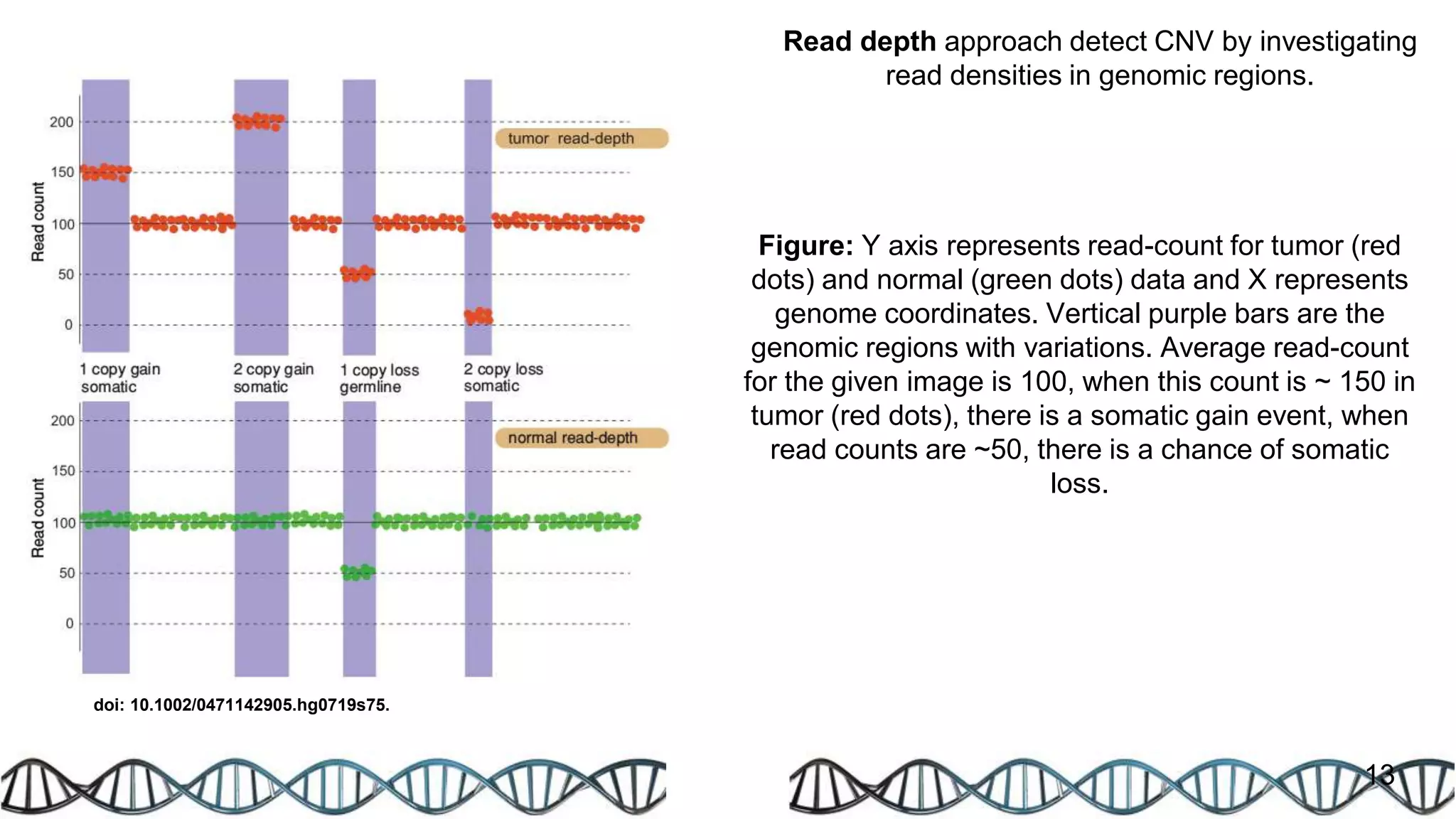
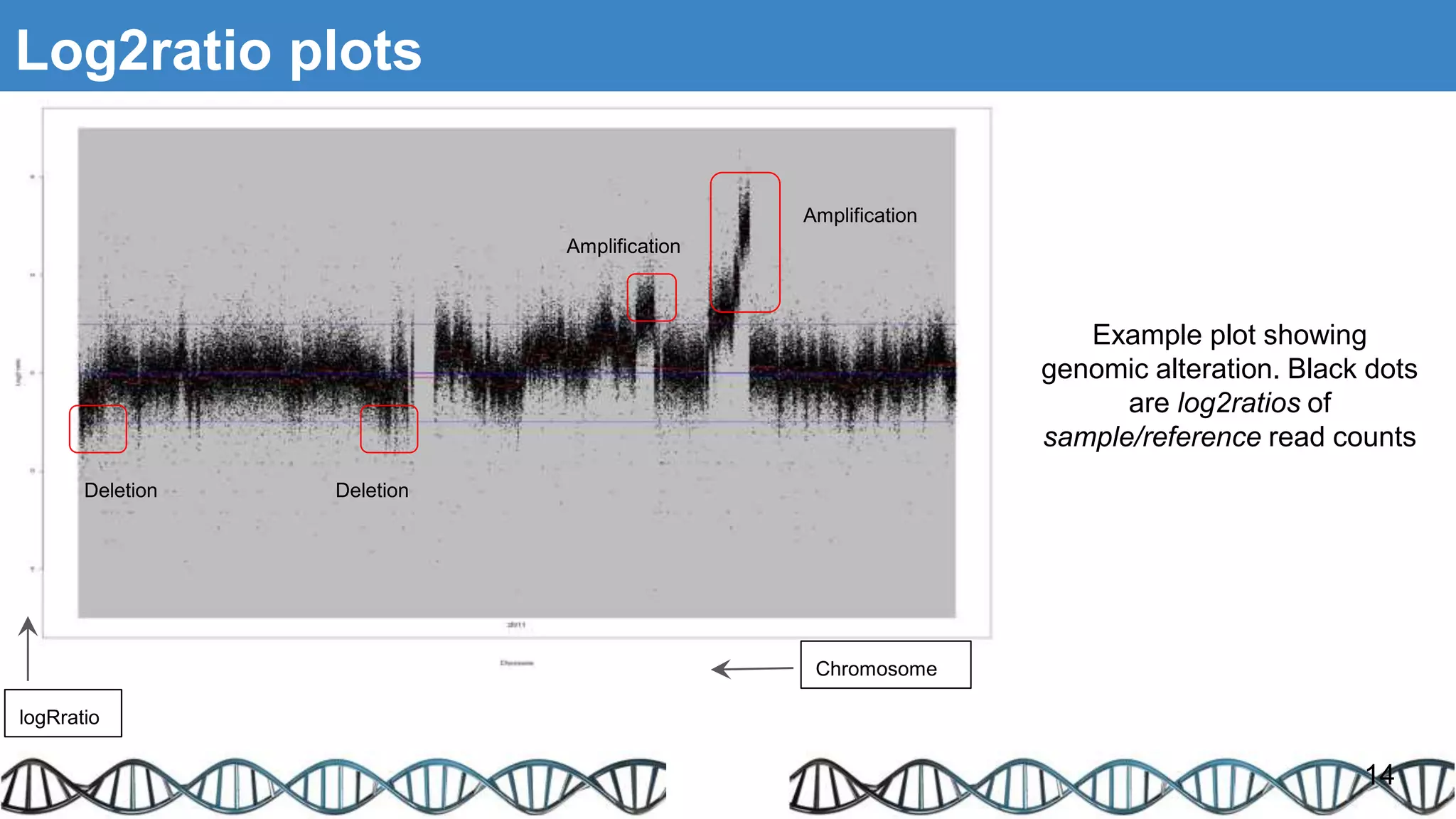
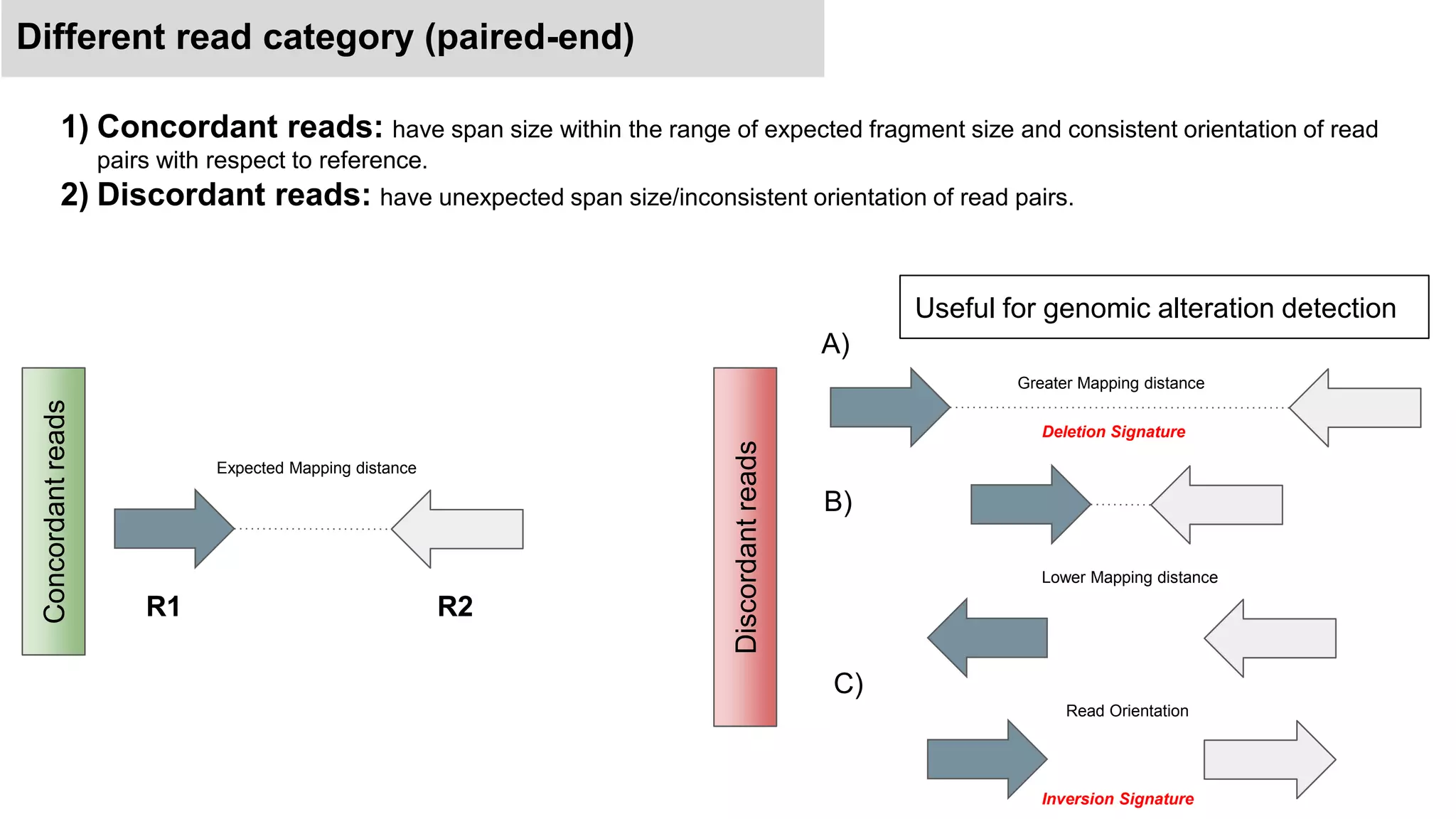
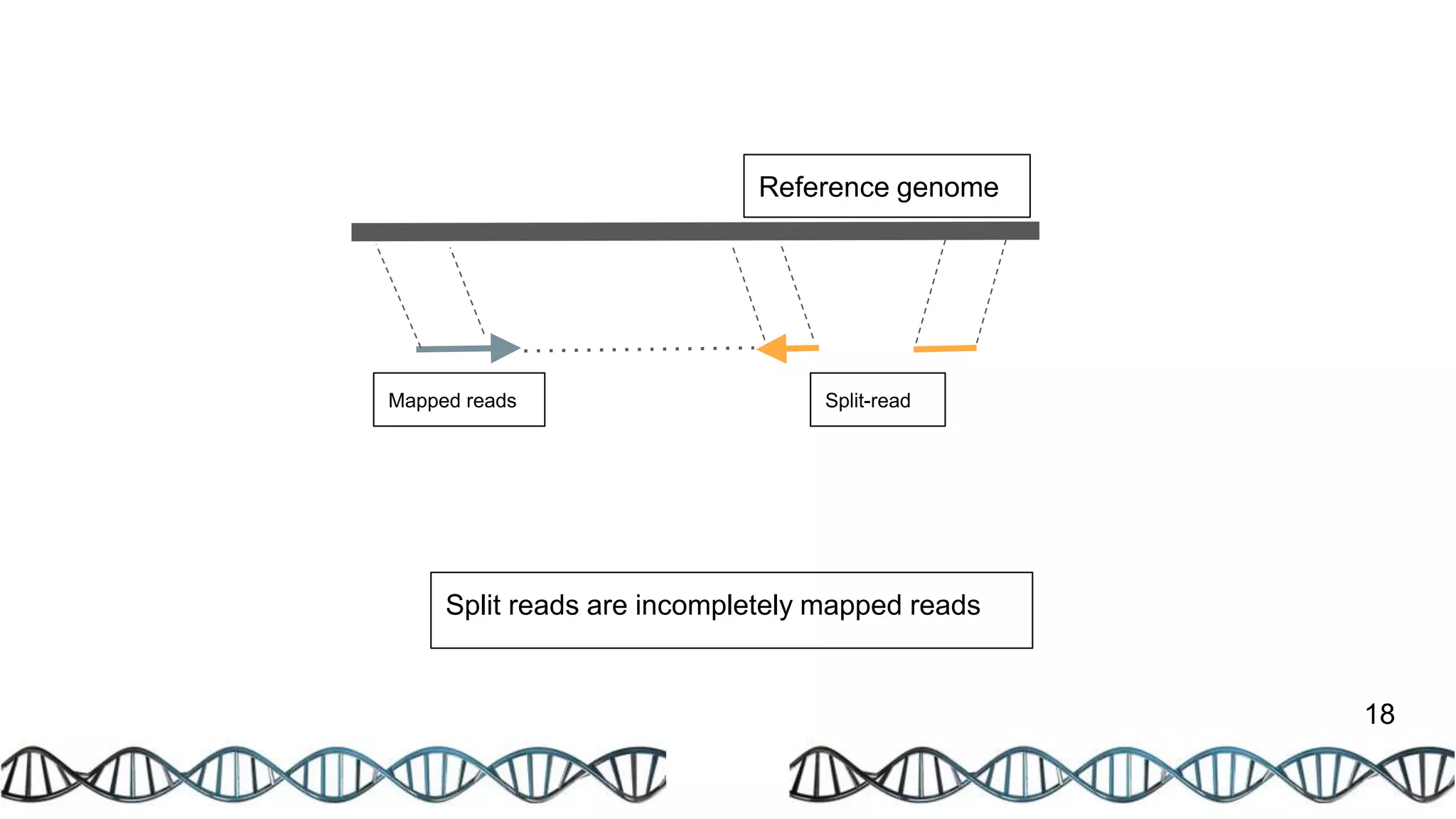



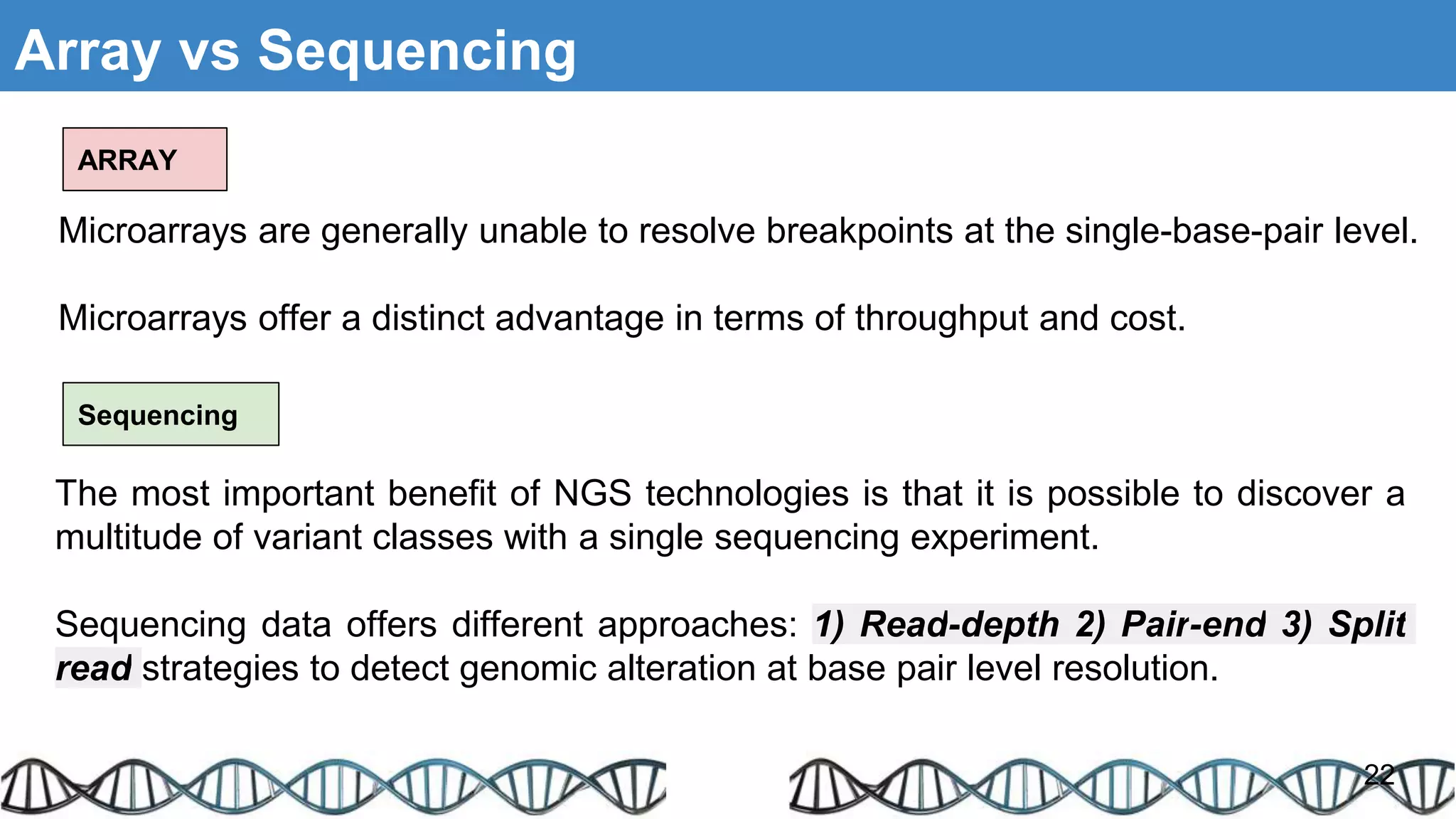



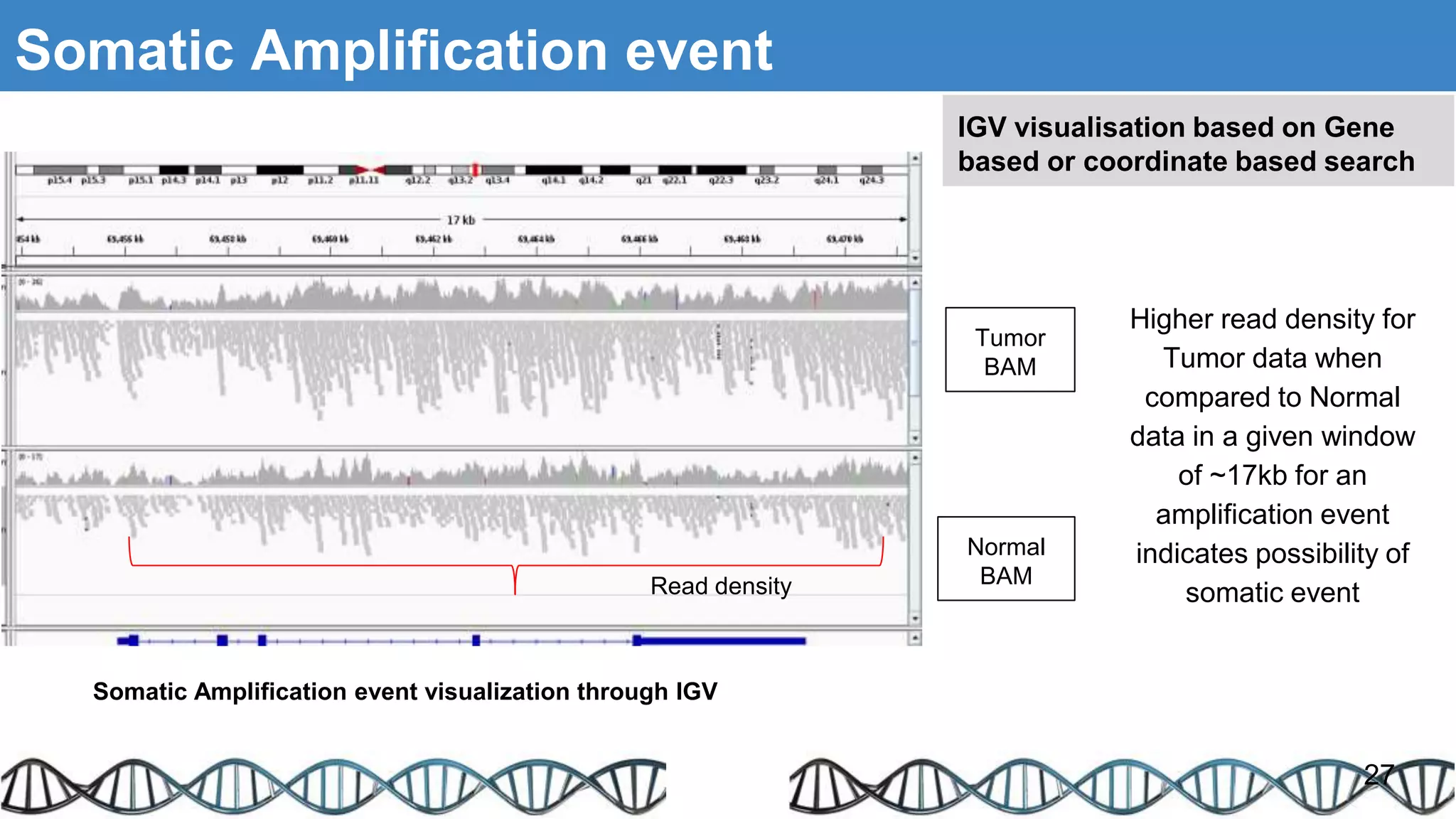
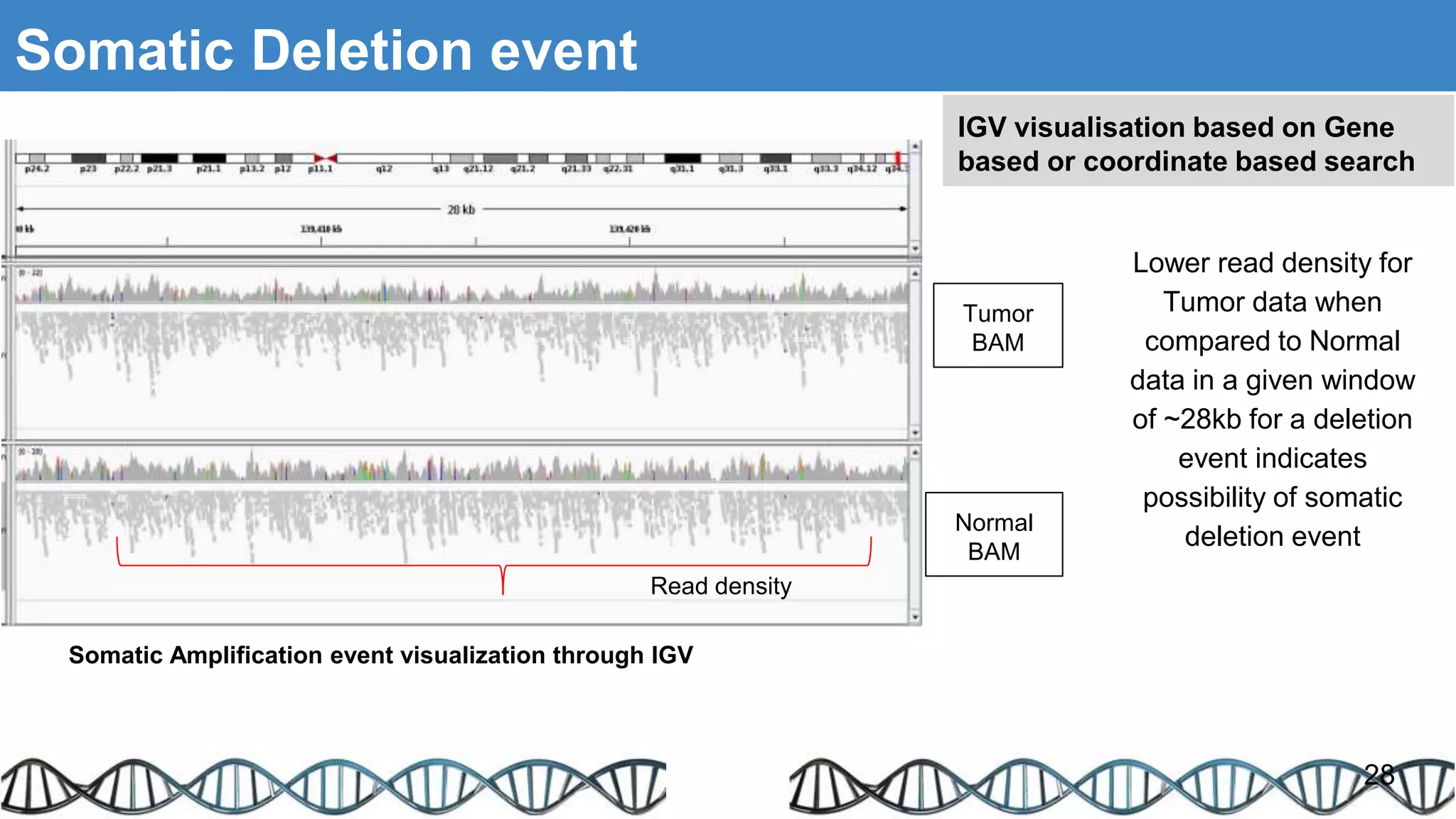
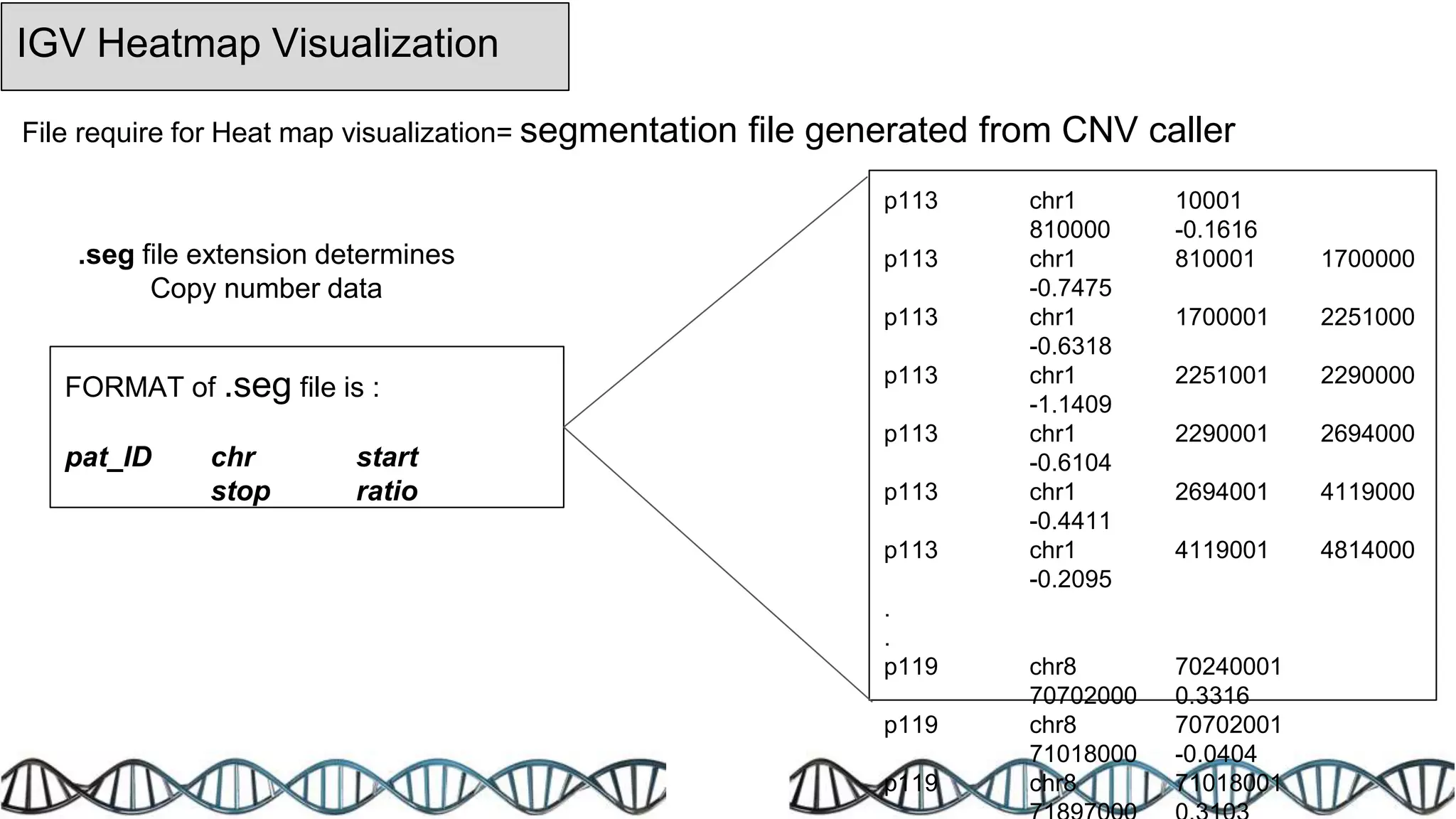
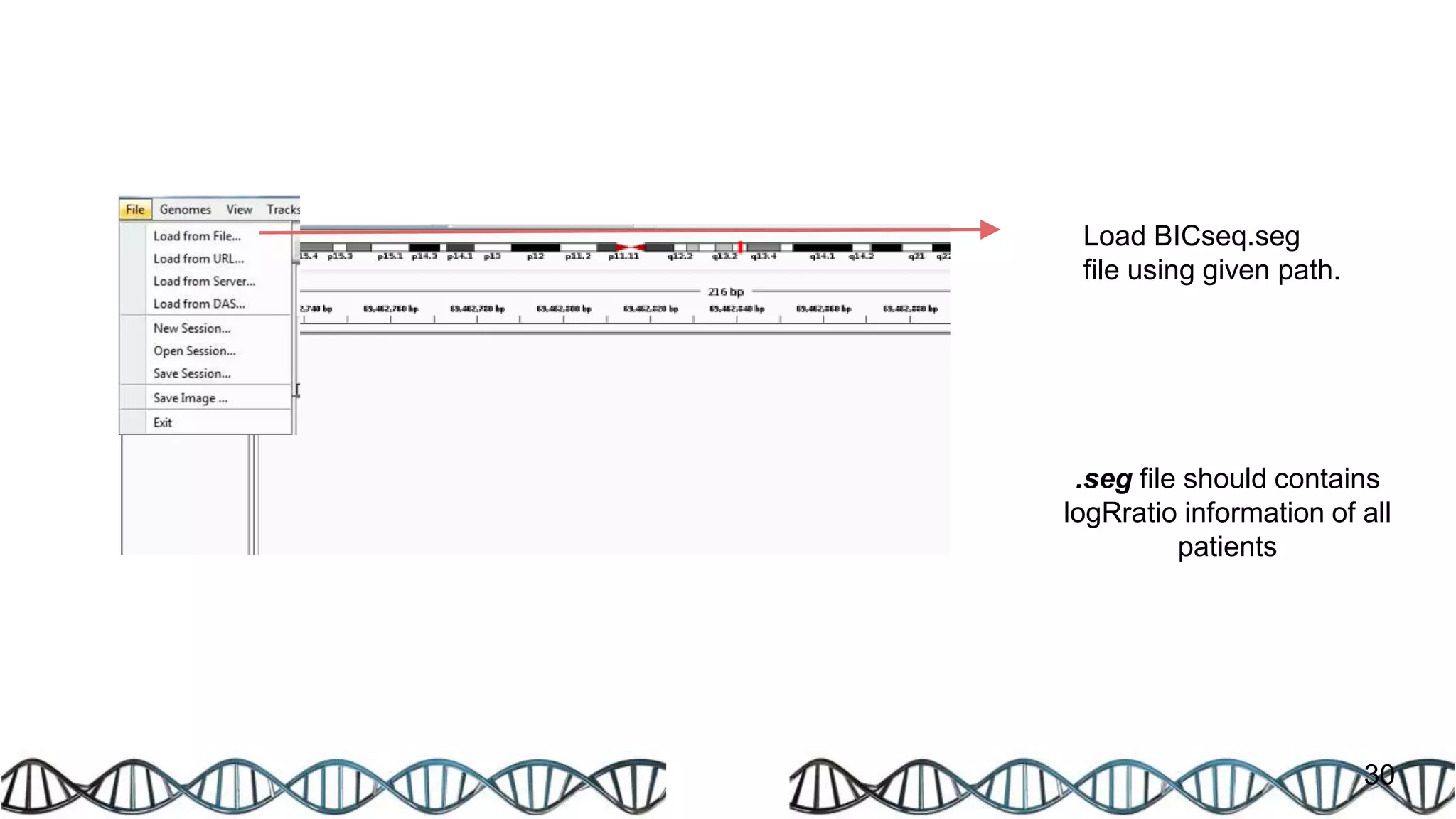

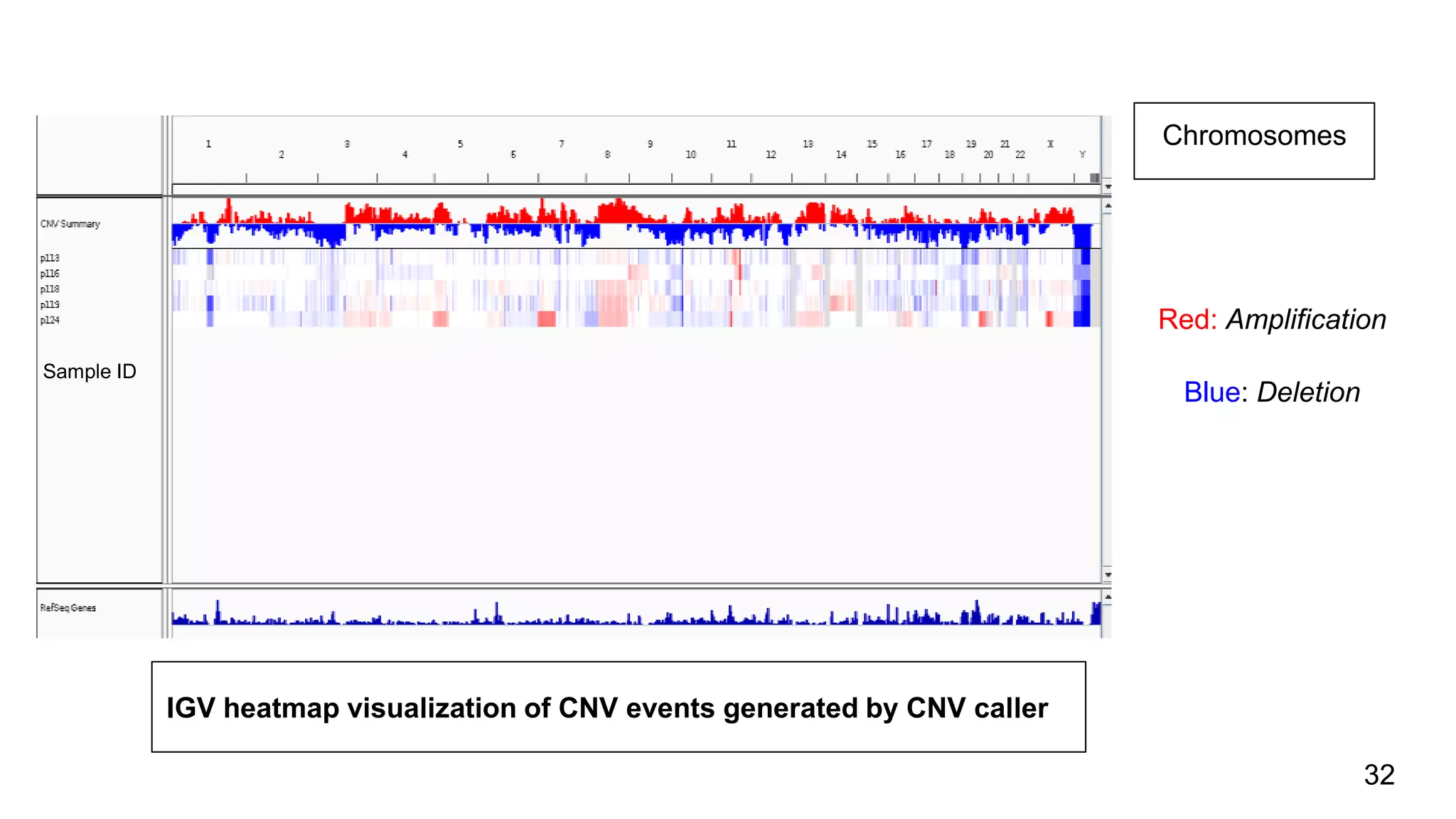


The document discusses genomic alterations identified through sequencing data, focusing on structural variations such as copy number variations (CNV), insertions, inversions, and translocations. It highlights various detection methods for these alterations, including array comparative genomic hybridization and next-generation sequencing (NGS) techniques, while emphasizing the advantages of NGS for precise characterization. The necessity for more robust algorithms is also noted, as current methods yield variable results in genomic alteration detection.


































































