Downloaded 28 times


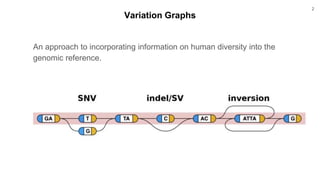
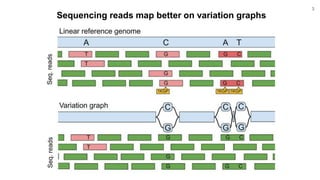
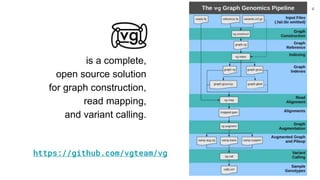
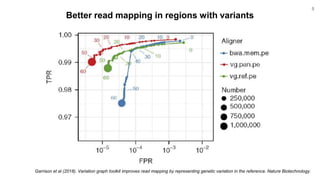
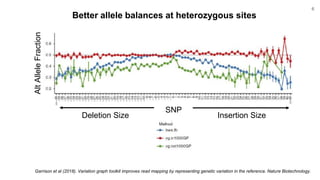
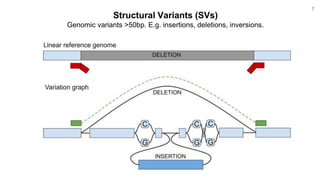
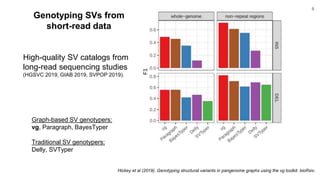
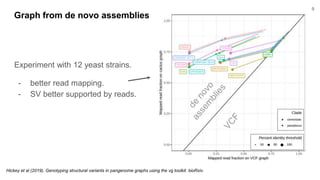
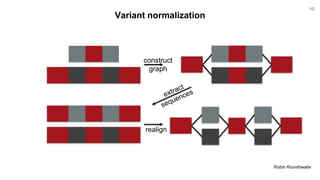
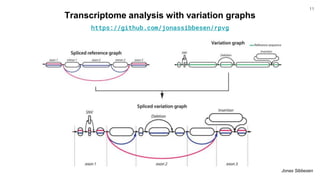
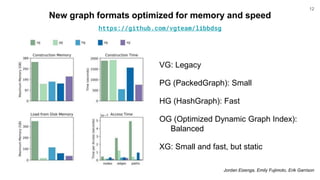






The document discusses updates to the vg toolkit for genome variation graphs, which enhance read mapping and variant calling by representing genetic diversity in genomic references. It reviews previous research on structural variant genotyping and introduces new graph formats and indexes for improved efficiency. The toolkit and associated resources are aimed at supporting better analysis and understanding of genomic variation.























































































