Downloaded 251 times








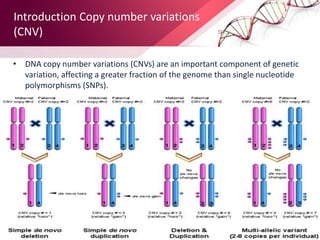



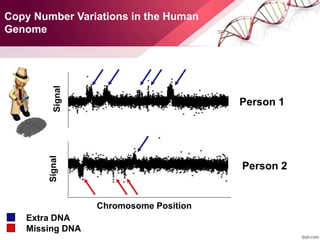



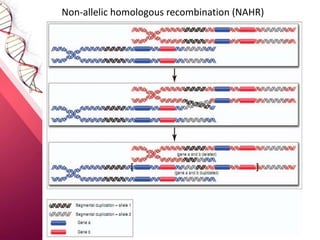
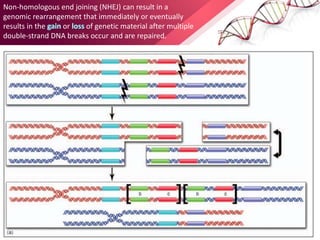
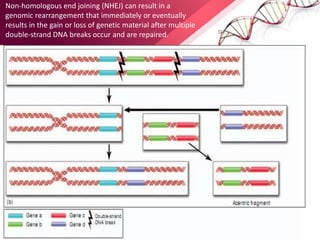











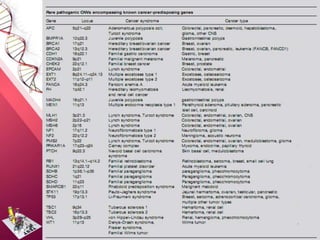
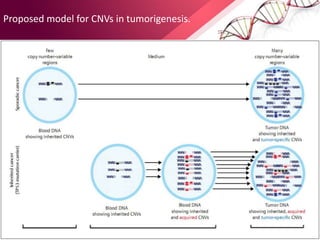




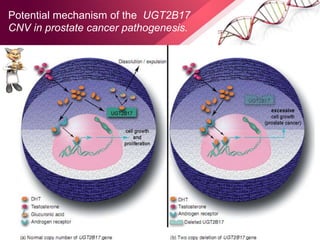
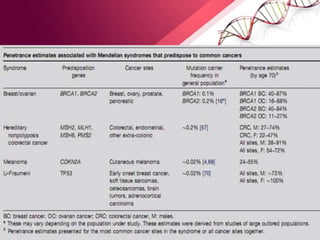

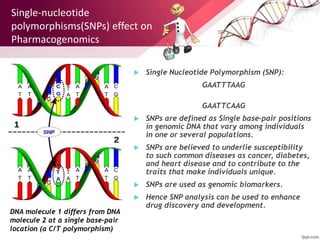
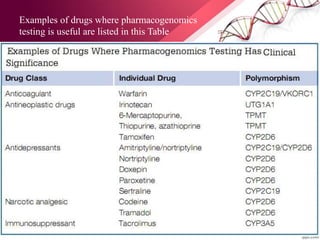


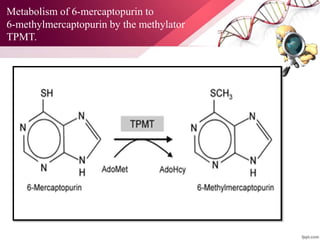

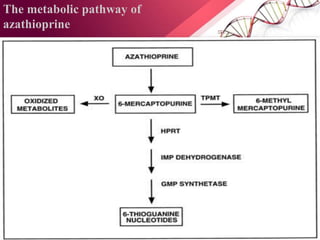
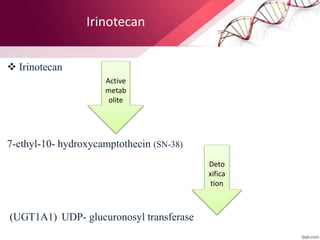
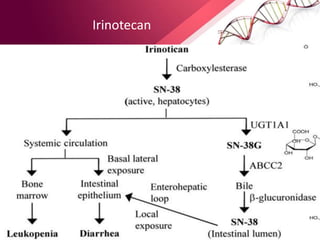
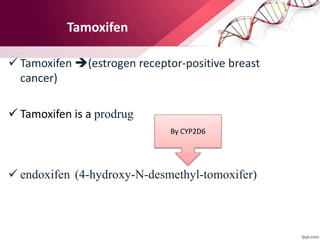









This document provides an agenda for a lecture on copy number variations (CNV) and their role in cancer development and pharmacogenetics. It begins with an introduction to CNVs, including definitions and mechanisms for their creation. It then discusses how CNVs can contribute to disease susceptibility and notes their role in directly influencing cancer cell genomes. The document outlines how common and rare CNVs may serve as "first hits" to the tumor genome or influence cancer risk. It provides some examples of specific cancers associated with CNVs and discusses how pharmacogenetics focuses on CNV effects in cancer treatment for drugs like tamoxifen and irinotecan. The document concludes by noting the promising potential for further discovery regarding C












![Cancer genetics [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/cancergeneticsautosaved-200614190344-thumbnail.jpg?width=640&height=640&fit=bounds)











































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

















