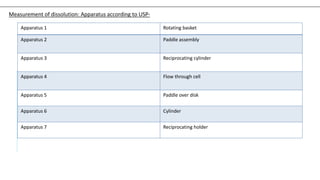
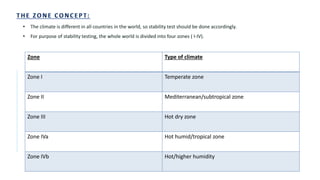
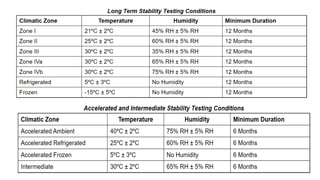
The document discusses the principles and methods of stability, solubility, pKa, and dissolution rate testing in pharmaceutical product development, emphasizing the importance of stability testing to ensure product quality and safety. It outlines various testing approaches such as real-time, accelerated, and retained stability testing, along with their objectives, acceptable conditions, and methodologies. Additionally, the document explains key concepts related to solubility and acid dissociation constant (pKa), which are critical for understanding drug formulation and behavior.










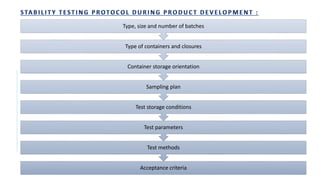

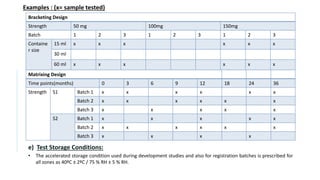
![f) Test Parameters:
• The stability test protocol should define test parameters that would define the test parameters that would be used for
evaluation of the stability samples.
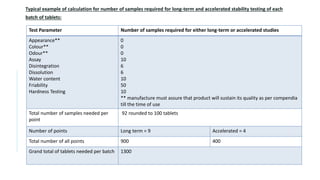
• Stability parameters for various types of products are listed [WHO, 2009]
DOSAGE FORM TEST PARAMETERS
Tablets Dissolution, Disintegration, Water content, Friability
Hard gelatin capsules Brittleness, Dissolution, Disintegration, Level of microbial contamination
Soft gelatin capsules Dissolution, Disintegration, Level of microbial contamination, pH
Oral Solutions,
Suspension and Emulsion
Clarity, Precipitate formation, pH, Viscosity, Density
Suspension : Dispersibility, Rheological properties, Distribution of particles
Emulsion : Phase separation, Mean size of dispersed globules
Powders and Granules for
Oral Solution, Suspension
Water content
Metered dose inhalers Dose content uniformity, Labelled number of medication actuations per
container meeting dose uniformity , aerodynamic particle size distribution,
microscopic evaluation, water content, leak rate, pump delivery, foreign
particulate matter
Nasal sprays: Solutions
and Suspension
Clarity, Level of microbial contamination, pH, particulate matter , Foreign
particulate matter
Suppositories Softening range, Disintegration and Dissolution time ( 37º)](https://image.slidesharecdn.com/pd-ad-240523130034-55325435/85/PD-Stability-pka-dissolution-ratw-pptx-16-320.jpg)




![• Ka or acid dissociation constant is a quantitative measurement of the strength of an acid in solution.
• Let us consider the dissociation of the compound ‘HA’ :
HA ⇌ A- + H+
The Ka for this reaction will be given by:
Ka = [A−][H+]
[HA]
• Expressing acidity in terms of Ka can be inconvenient for practical purposes, therefore, pKa is used.
• pKa can be defined as ‘the negative base-10 logarithm of acid dissociation constant (Ka) of a solution’.
pKa = -log10Ka
Example:
The Ka constant for acetic acid is 0.0000158, but the pKa constant is 4.8, which is a simpler expression.
The smaller the pKa value, the stronger the acid.
The pKa value of lactic acid is about 3.8, so that means lactic acid is stronger than acetic acid.](https://image.slidesharecdn.com/pd-ad-240523130034-55325435/85/PD-Stability-pka-dissolution-ratw-pptx-25-320.jpg)

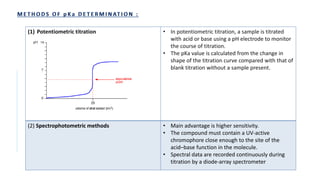
![• Requirements- Aspirin, ethanol, sodium hydroxide, pH meter.
• Principle- Aspirin is a weak acid and partially ionizes in water.
• HA + H2O ↔ H3O+ + A-
It’s acid dissociation constant, Ka is given by:
Ka = [A−][H3O+]
[HA]
Aspirin and sodium hydroxide react in a 1:1 mole ratio:
Method-
1. A burette is filled with 0.1M sodium hydroxide solution.
2. 0.36g of aspirin is weighed in 250ml beaker and 10ml of 95% ethanol is added and volume is made up with deionized water.
3. 2ml portions of sodium hydroxide solution is added from burette to the beaker, stirring well between each additions and recording the pH using a pH
meter.
4. The pH begins to rise rapidly near the end-point
5. After adding 18ml of sodium hydroxide solution, addition is continued in 0.5ml portions.
6. After adding about 22ml, additions in 2ml portions is started again.
7. The addition is continued until total of 36ml has been added.
8. A Graph of pH against volume of 0.1M sodium hydroxide is plotted.
9. The end-point is calculated from the graph of titration.](https://image.slidesharecdn.com/pd-ad-240523130034-55325435/85/PD-Stability-pka-dissolution-ratw-pptx-29-320.jpg)