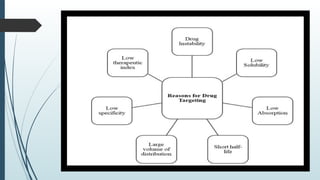
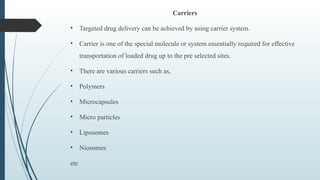
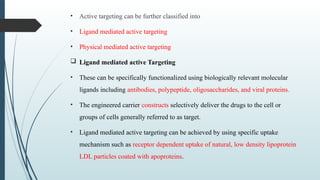
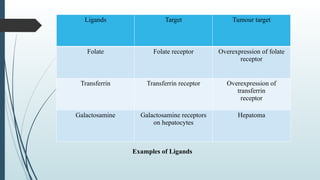
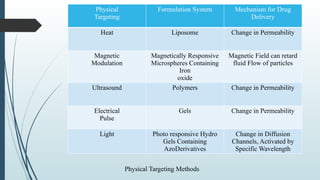
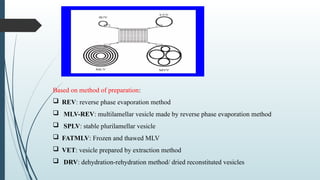
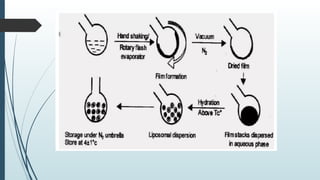
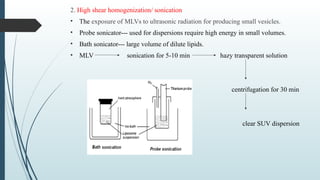
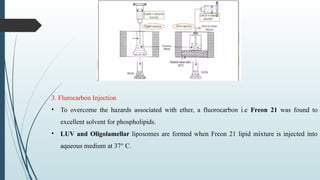
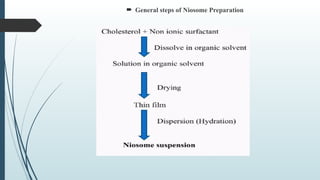
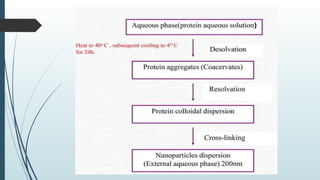
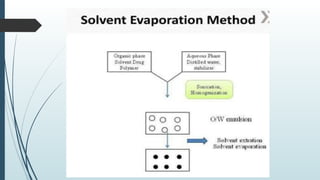
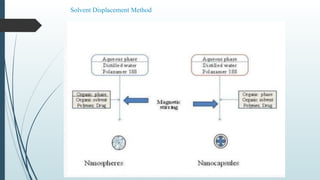
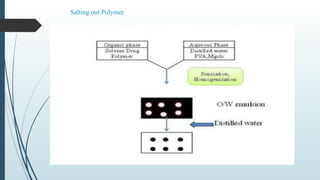
The document provides a comprehensive overview of targeted drug delivery systems, detailing their concepts, advantages, disadvantages, and various strategies, including the use of different carriers like liposomes and nanoparticles. It emphasizes the importance of targeting drugs to specific sites to enhance therapeutic efficacy while reducing side effects. Additionally, it discusses methodologies for drug targeting, such as passive, active, and double targeting, along with examples and classifications of carriers and their preparation methods.























































































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)

















