Downloaded 456 times

















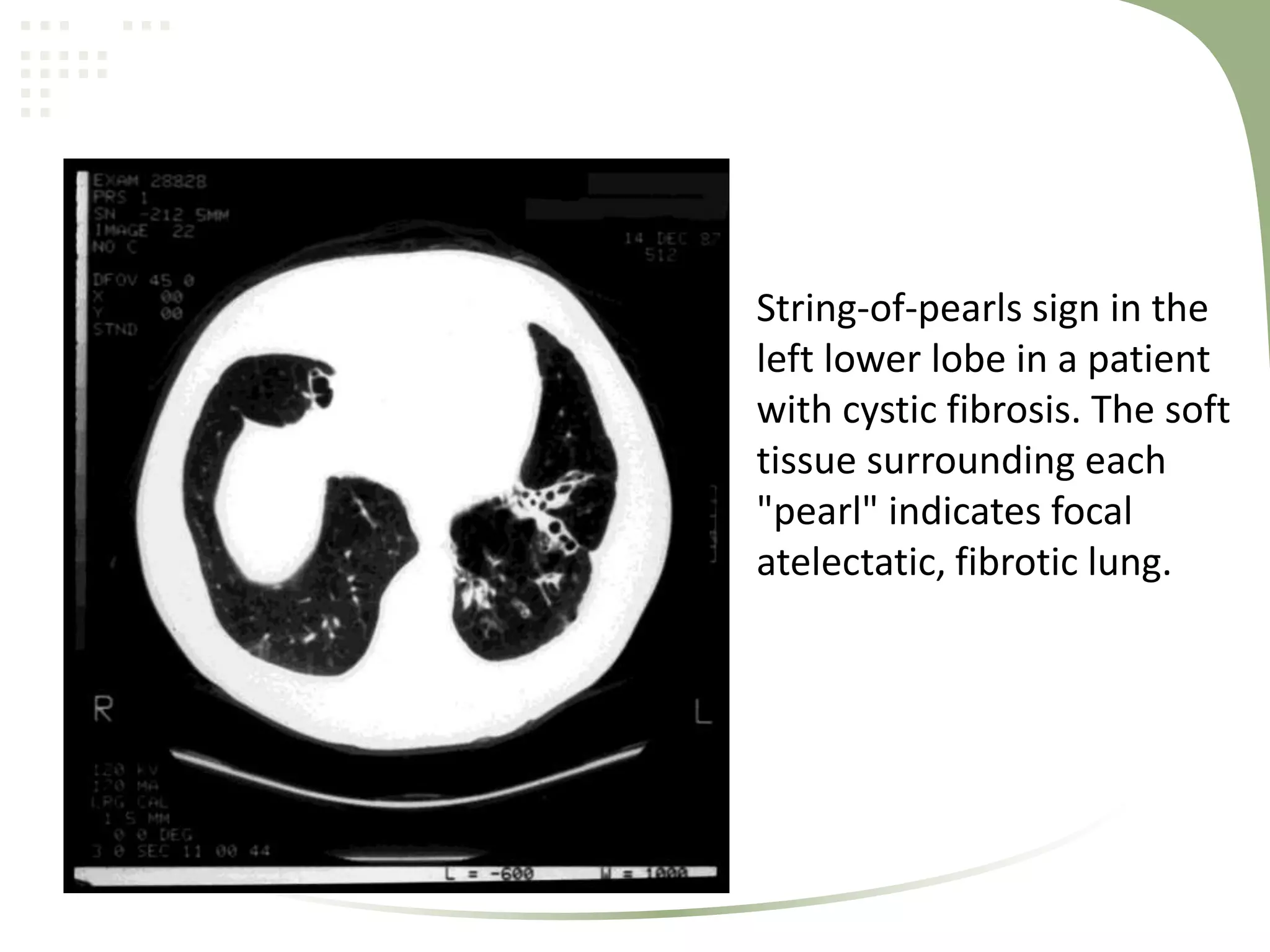
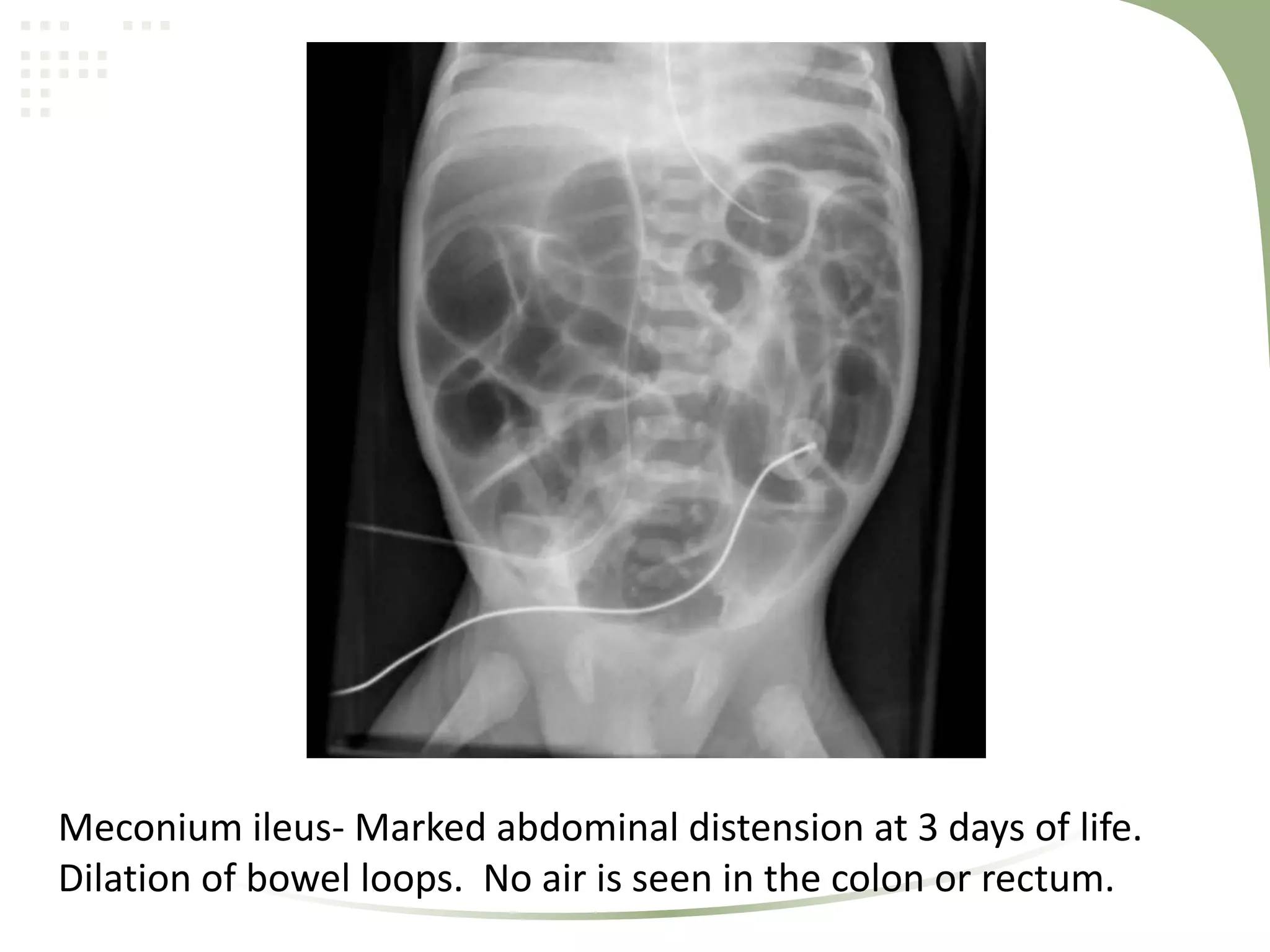















Cystic fibrosis is an inherited disease affecting the lungs and digestive system. It is caused by mutations in the CFTR gene, which encodes a chloride channel. This leads to thick, sticky mucus buildup in the lungs, pancreas, and other organs. Symptoms include persistent lung infections, gastrointestinal issues like poor growth. The diagnosis is made through genetic testing, sweat tests, and clinical features. Treatment focuses on airway clearance, antibiotics, enzymes, vitamins, and managing complications through medications, airway therapy, exercise, and sometimes surgery or lung transplantation.























































































