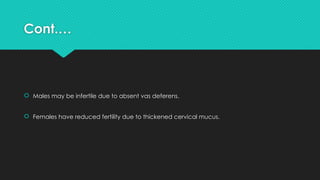
Cystic fibrosis is a chronic genetic disorder caused by mutations in the CFTR gene, leading to severe respiratory and digestive issues, primarily due to thickened secretions and mucus. Common symptoms include salty skin, poor weight gain, and recurrent lung infections, with the delta F508 mutation being the most prevalent. Treatment focuses on managing symptoms and complications through antibiotics, inhaled medications, and airway clearance techniques to improve quality of life.