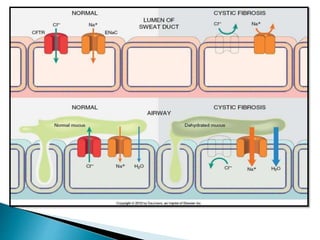
Cystic fibrosis is an inherited disorder that affects the lungs and digestive system. It is caused by mutations in the CFTR gene that result in thick, sticky mucus buildup in organs. Symptoms include persistent lung infections, problems digesting food, and other issues. Treatment focuses on loosening mucus, treating infections, improving nutrition, and managing symptoms, though there is no cure currently. Screening at birth and ongoing testing monitors disease progression and treatment effectiveness.



































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)



