Motif finding plays a vital role in identifying transcription factor binding sites (TFBS) that help understand gene expression regulation. The document discusses various approaches to motif finding including:
1. Enumerative approaches that search for consensus sequences like YMF, DREME, CisFinder, Weeder, FMotif, and MCES.
2. Probabilistic approaches using position weight matrices (PWM) and algorithms like EM, MEME, STEME, EXTREME, Gibbs sampling, AlignACE, and BioProspector.
3. Other approaches like nature-inspired algorithms, combinatorial methods, and the LOGOS algorithm based on Bayesian modeling.
The document provides details on the methodology,







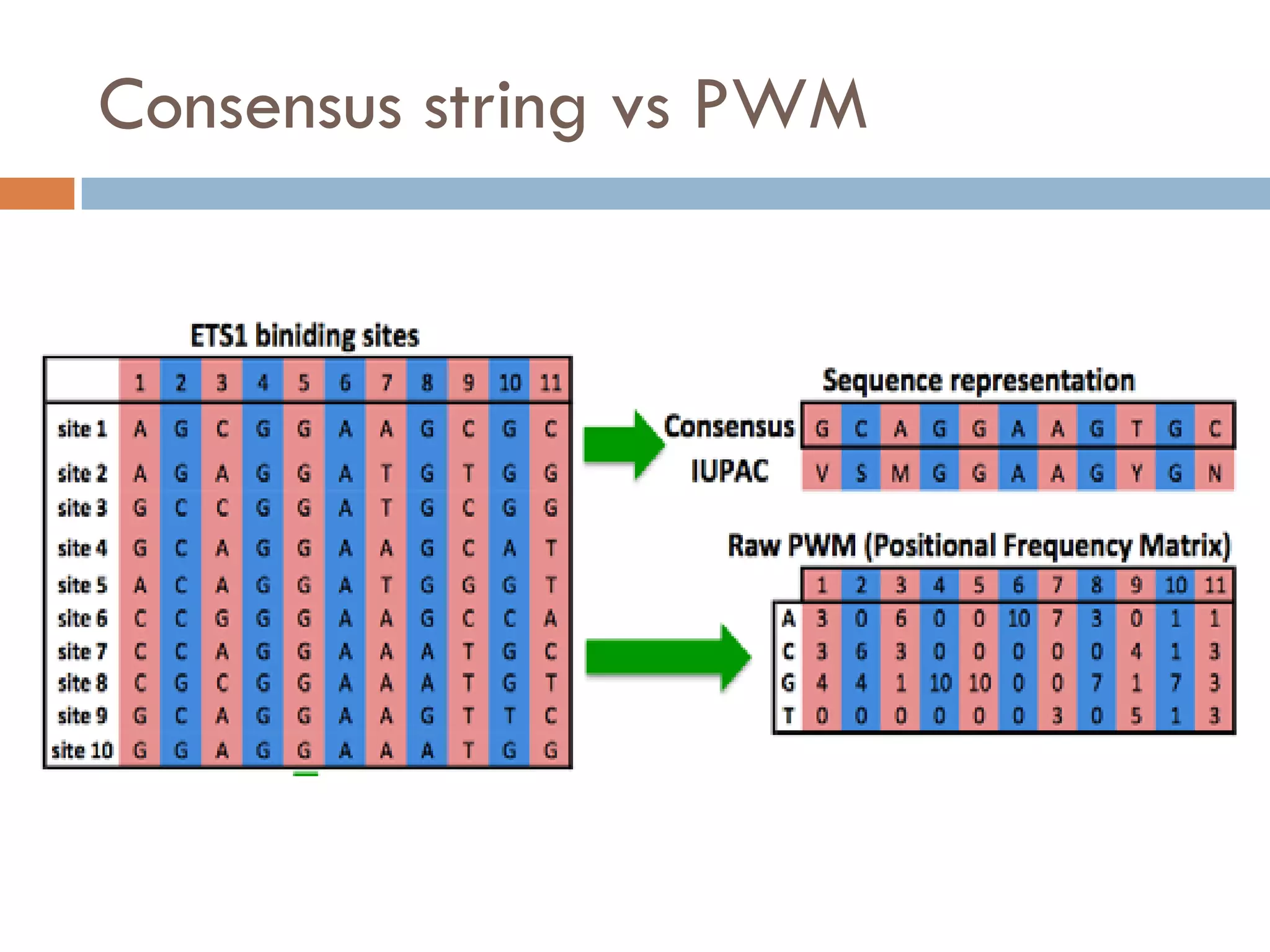
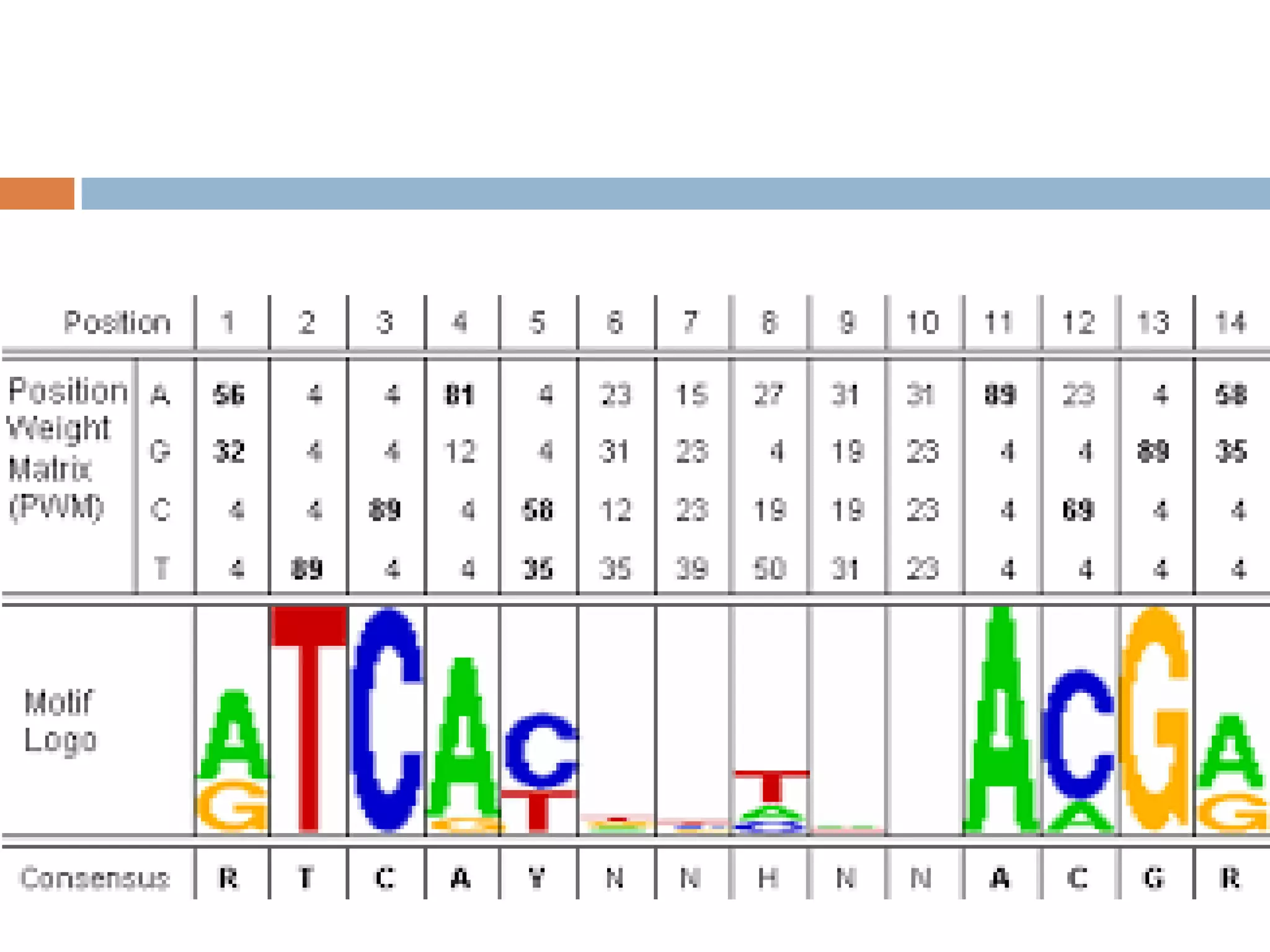



















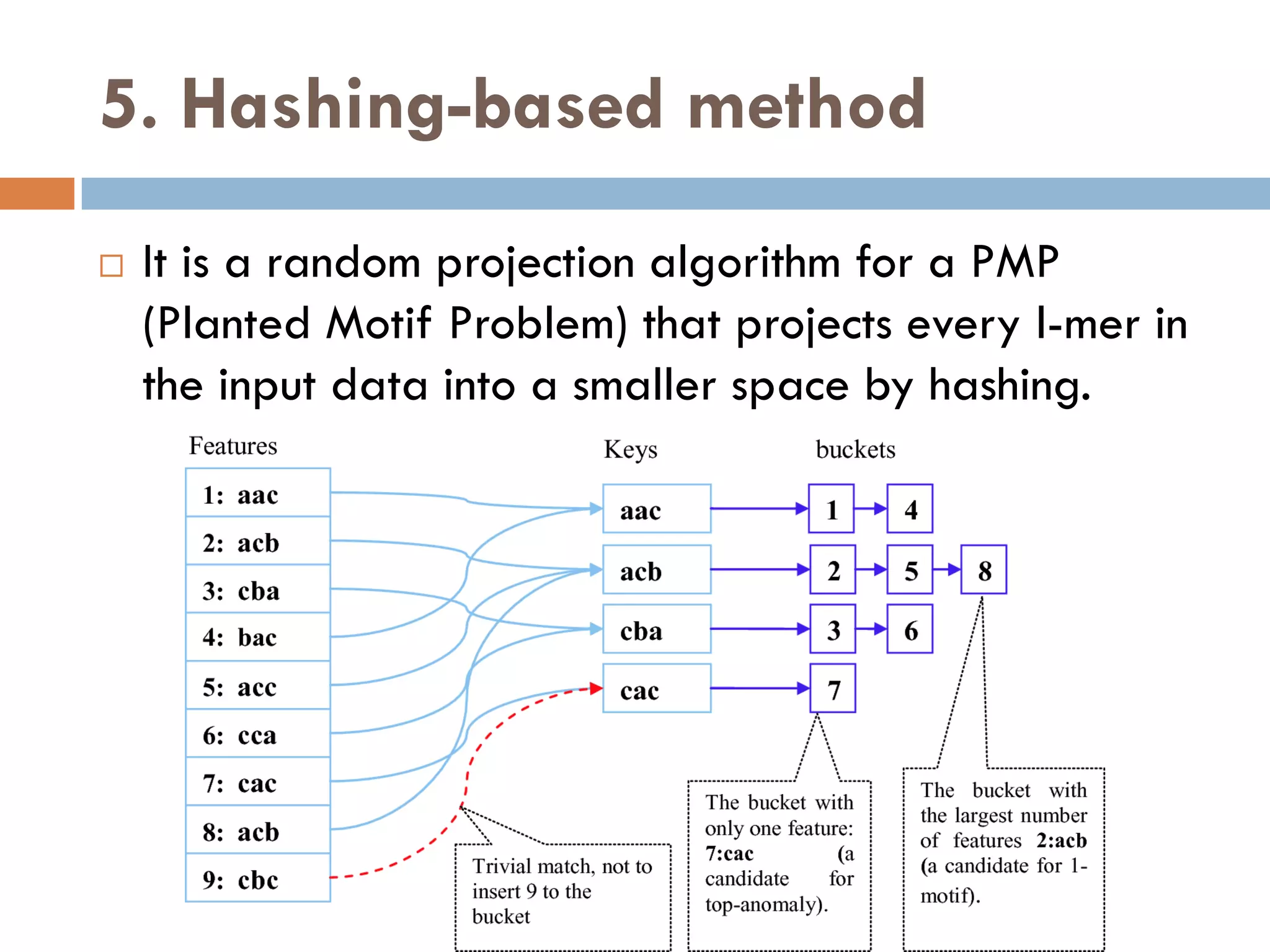


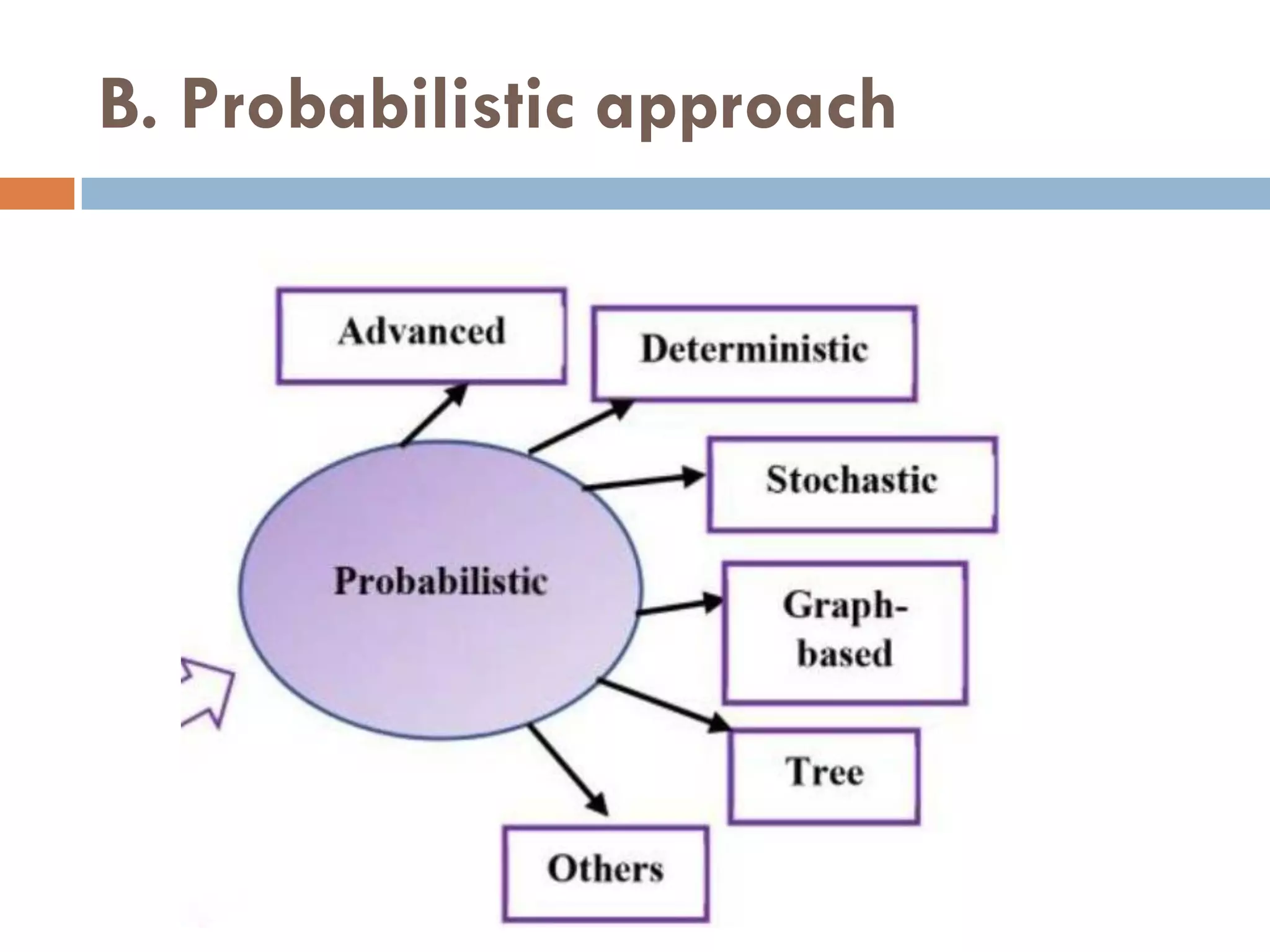



















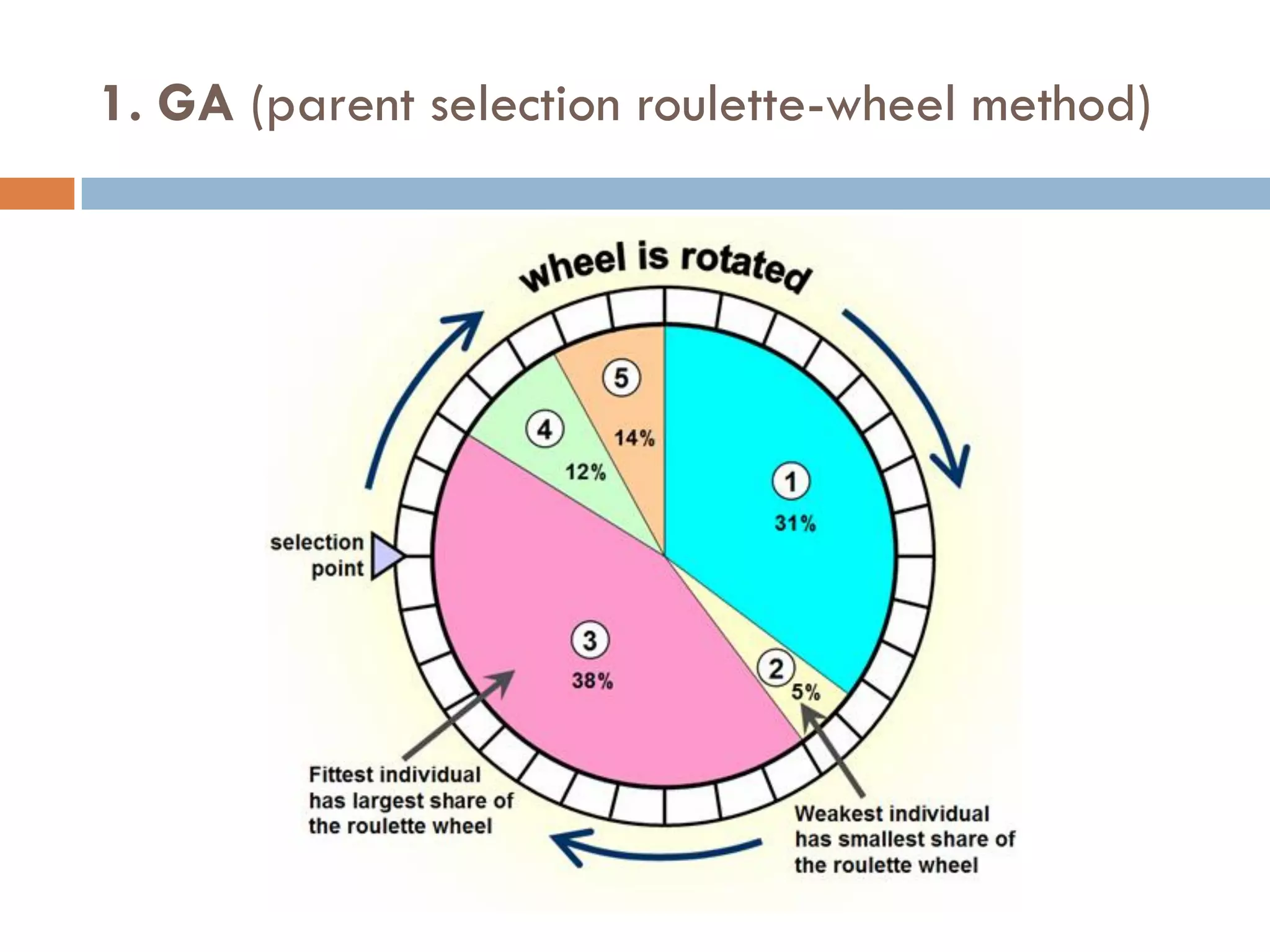















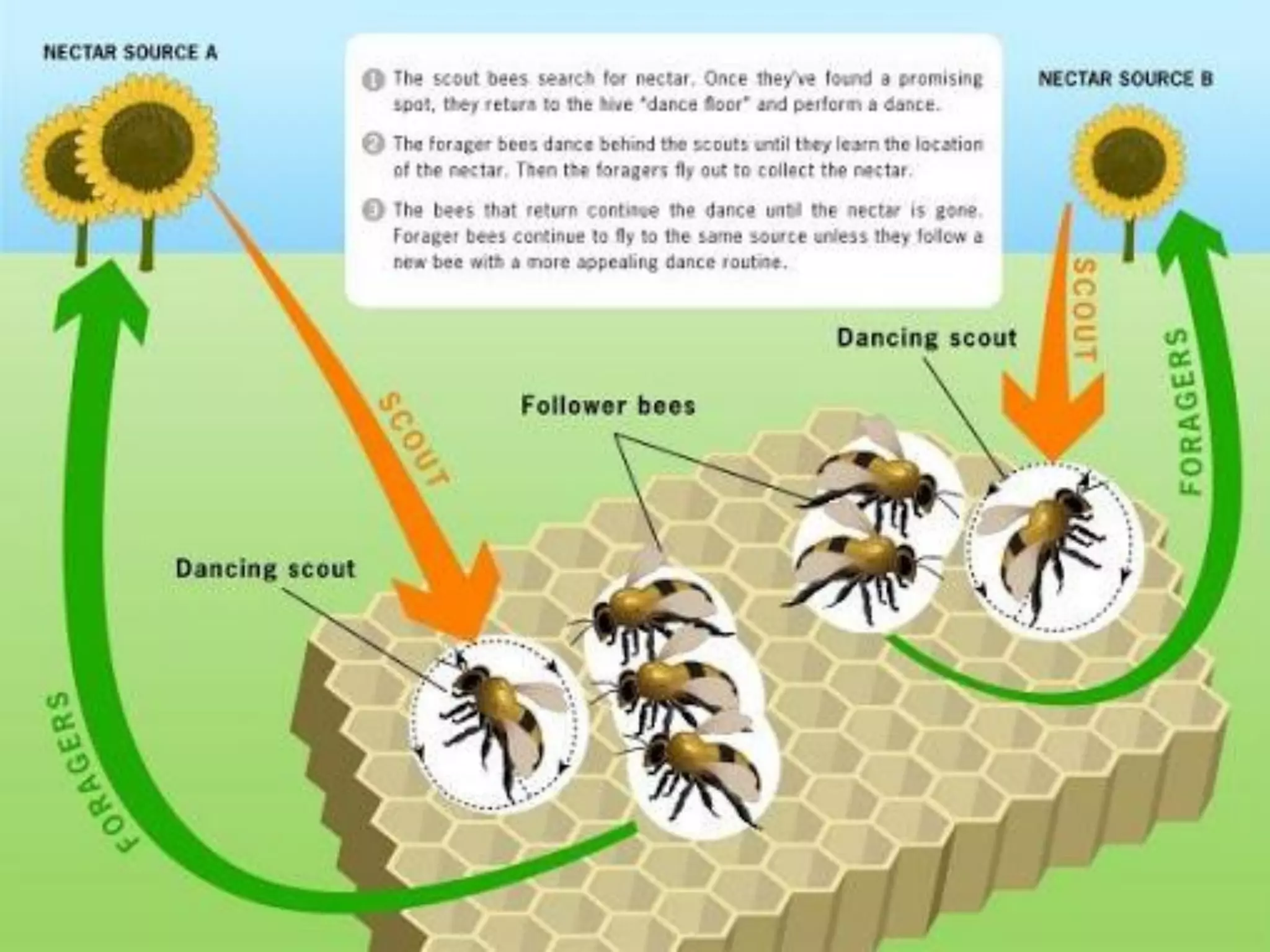
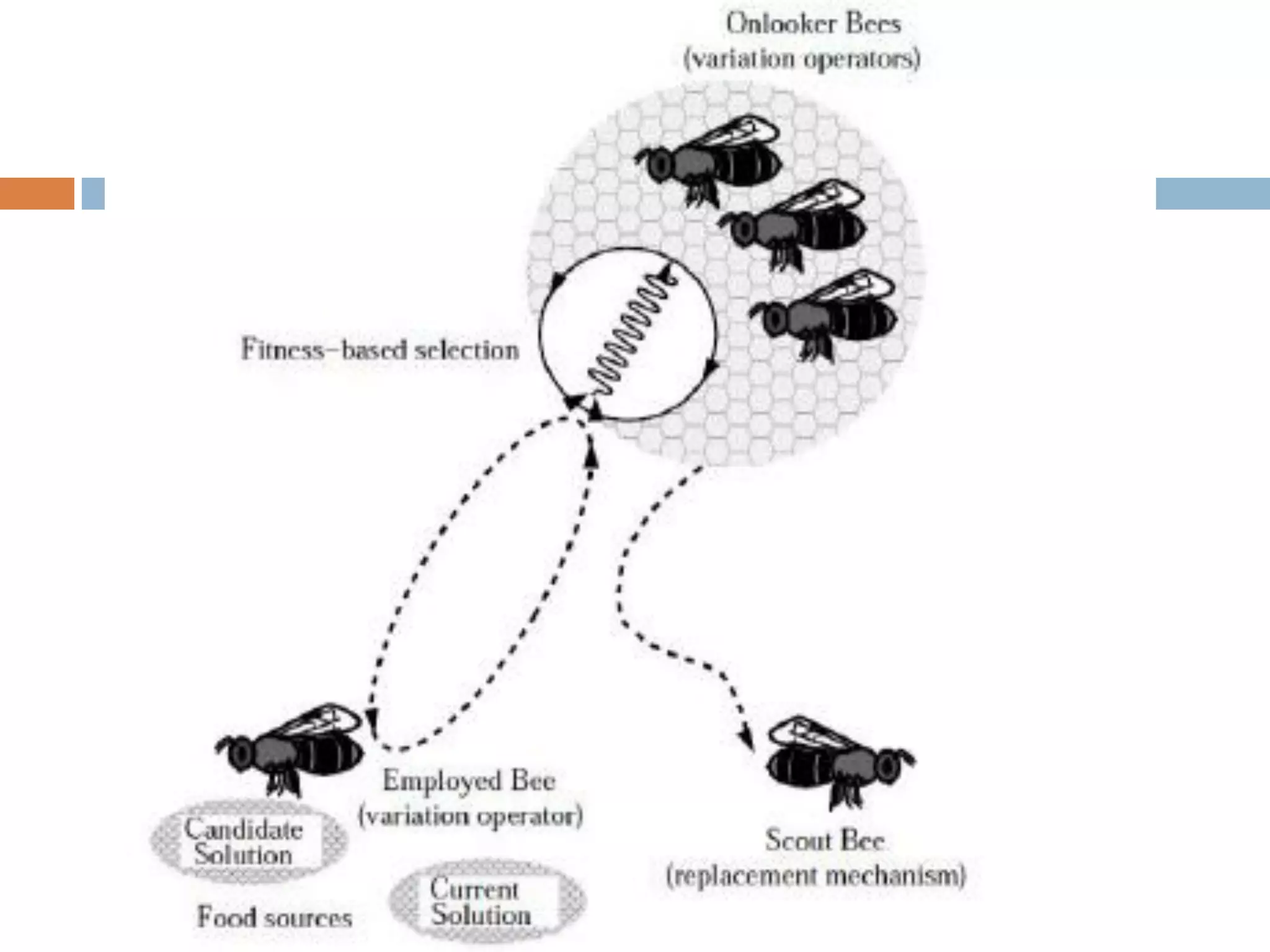



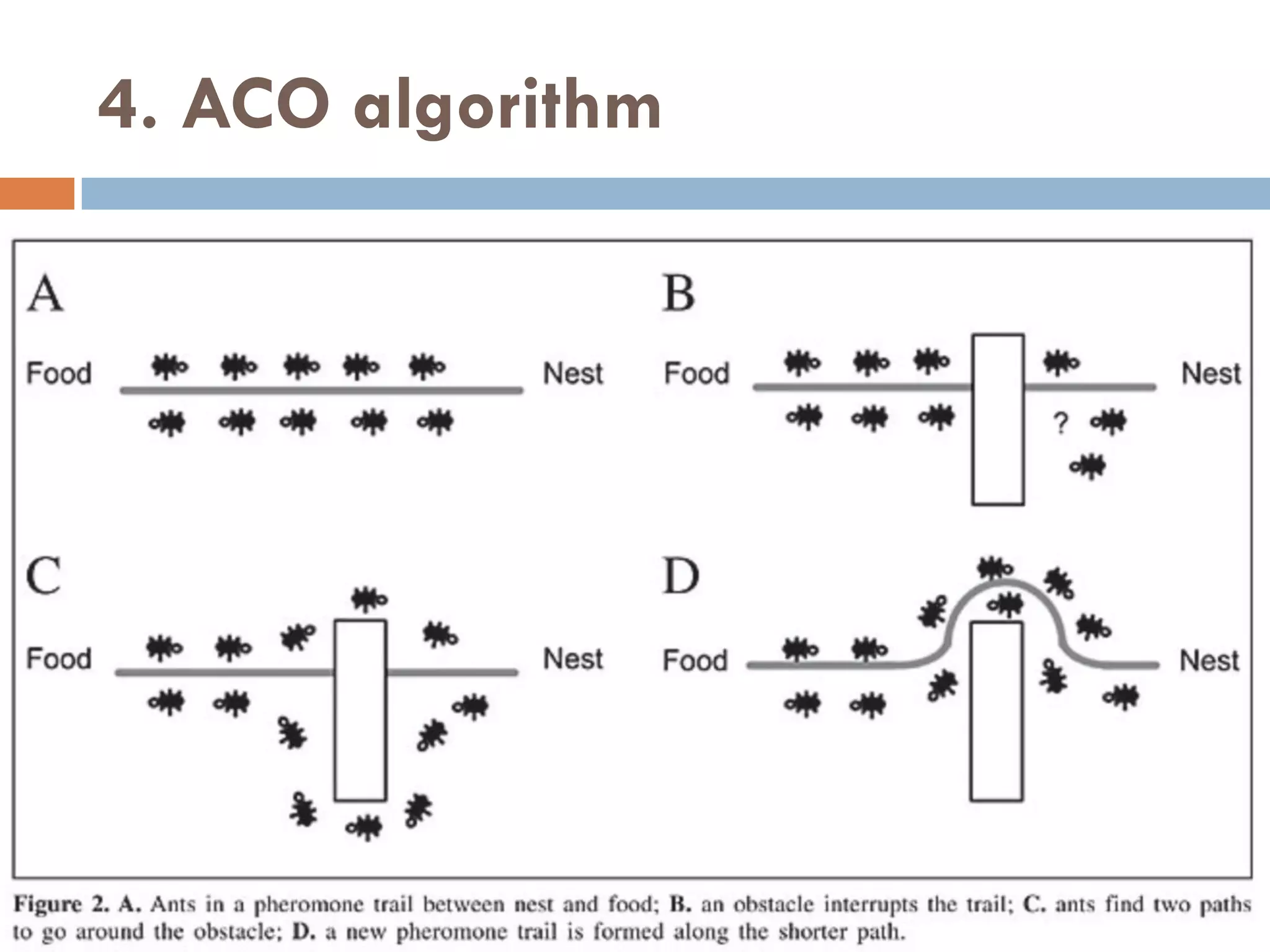



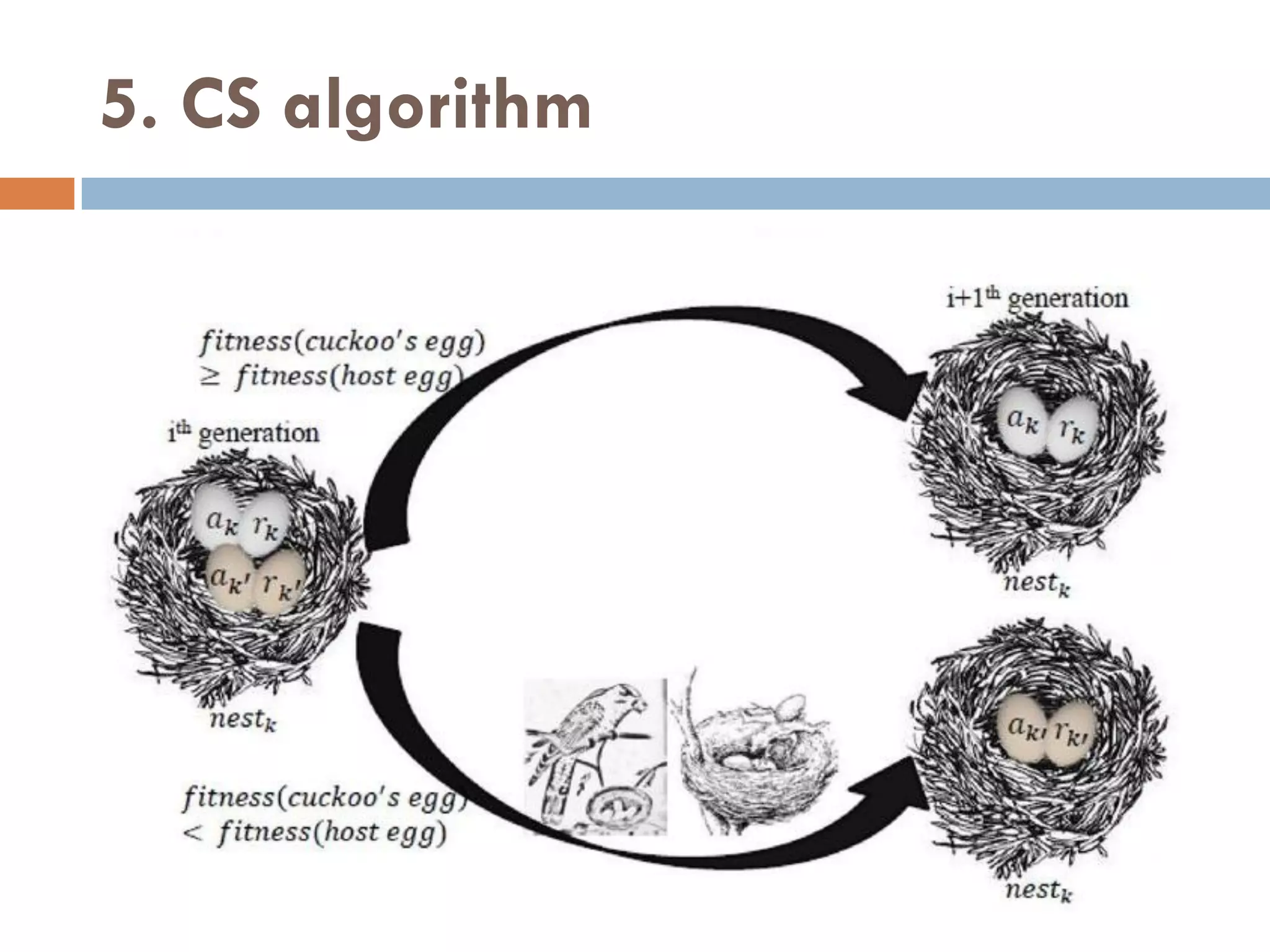
![5. CS algorithm
CS is a new simple heuristic search algorithm that is more efficient
than GA and PSO.
CS is inspired from brood parasitism reproduction behavior of
some cuckoo species in combination with Lévy flight behavior.
The cuckoos lay their eggs in nests of the other birds with the
abilities of selecting the lately spawned nests and removing existing
eggs to increase the hatching probability of their eggs.
If host birds discover these eggs, they either throw them away or
abandon the nest and build a new nest.
CS is characterized by the subsequent rules:
1. Each cuckoo lays one egg at a time and disposes its egg in a
random selected nest.
2. The nests that have high quality of eggs (Solutions) are the best
and will continue to the following generations.
3. The number of accessible host nests is fixed, and a host bird can
discover a parasitic egg with a probability Pa E [0,1].](https://image.slidesharecdn.com/motiffinding-221031104130-3b7420f8/75/Motif-Finding-pdf-77-2048.jpg)










































![Mini_project_ppt_final[1].pptxwjwjwjishwbwhajjwjwjwiwjwj](https://cdn.slidesharecdn.com/ss_thumbnails/miniprojectpptfinal1-241129055447-ea75a1b8-thumbnail.jpg?width=640&height=640&fit=bounds)




























![[DSC Europe 25] Uros Pesic - The Reality of AI in Marketing.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/rtkodnmtycovsllvzsyn-9-251215095918-b0c6bfe3-thumbnail.jpg?width=640&height=640&fit=bounds)


![[DSC Europe 25] Dusan Nesic - Securing Tomorrow’s Infrastructure: Why Cyber-P...](https://cdn.slidesharecdn.com/ss_thumbnails/qikbszfftyowjm2q6duw-1-251211083848-8f2ead6b-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DSC Europe 25] Nikolay Burlutskiy - Best Practices for Building Enterprise M...](https://cdn.slidesharecdn.com/ss_thumbnails/uirvaiuvq8y1w8hzd9tx-7-251212103249-2619edb4-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DSC Europe 25] Kaja Kandare - LLM as a judge.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/arxyccaxsdsd1ba99wjw-7-251212104007-2b4e3f64-thumbnail.jpg?width=640&height=640&fit=bounds)

![[DSC Europe 25] Behzad Hosseini - AI Agents in the Wild: Deploying Models tha...](https://cdn.slidesharecdn.com/ss_thumbnails/3qtejajvsjqrzwfept2c-10-251212103250-7f2b1068-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DSC Europe 25] Branko Urosevic -Rethinking Financial Talent: Integrating Cod...](https://cdn.slidesharecdn.com/ss_thumbnails/8jjrus8ttko6qj64f58f-3-251212103250-642c6374-thumbnail.jpg?width=640&height=640&fit=bounds)


![[DSC Europe 25] Debmalya Biswas - Agentification: the art of transforming man...](https://cdn.slidesharecdn.com/ss_thumbnails/r5azlggvtqiaiiusrqdr-4-251212103249-5a12c89b-thumbnail.jpg?width=640&height=640&fit=bounds)


![[DSC Europe 25] Milan Sekuloski - Data, Defence, and Development: Cybersecuri...](https://cdn.slidesharecdn.com/ss_thumbnails/dfrkwwx4qly6atqpbl4z-4-251209104645-c3d4b0ca-thumbnail.jpg?width=640&height=640&fit=bounds)

