Download as PDF, PPTX




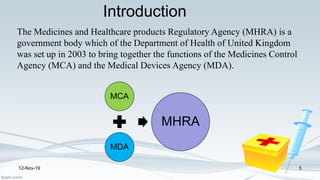








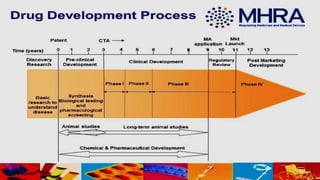
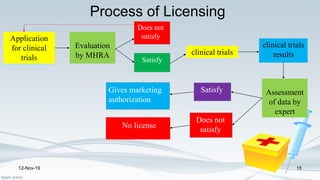


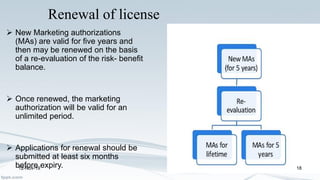











The Medicines and Healthcare products Regulatory Agency (MHRA) regulates medicines, medical devices and blood components for transfusion in the UK. It was formed in 2003 by merging the Medicines Control Agency and Medical Devices Agency. The MHRA licenses pharmaceutical companies, importers, clinical trials and new medicines. It oversees the drug development process from discovery through clinical trials to licensing. The MHRA also monitors drug safety after approval and can renew, cancel or amend licenses as needed.























































































