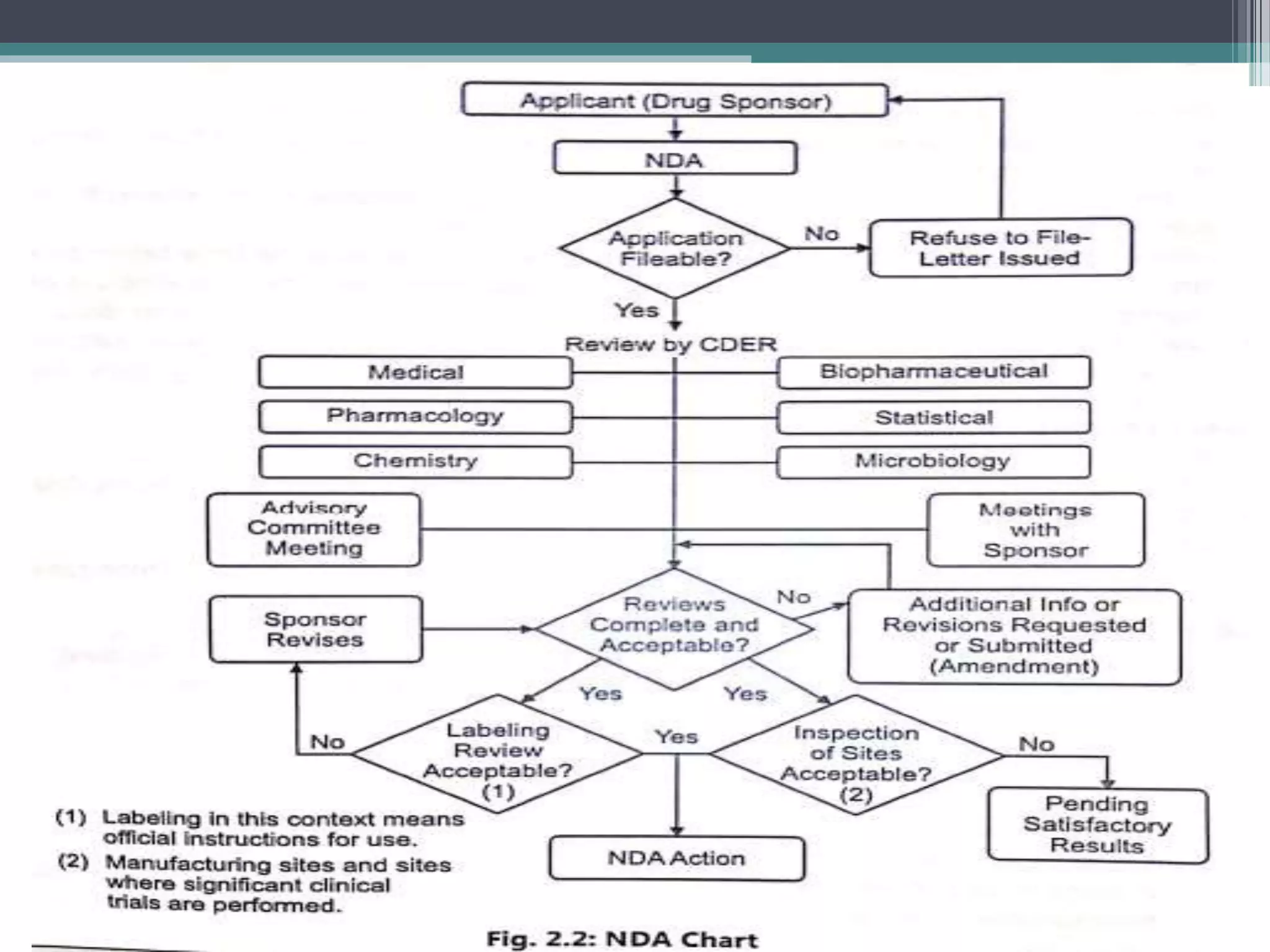
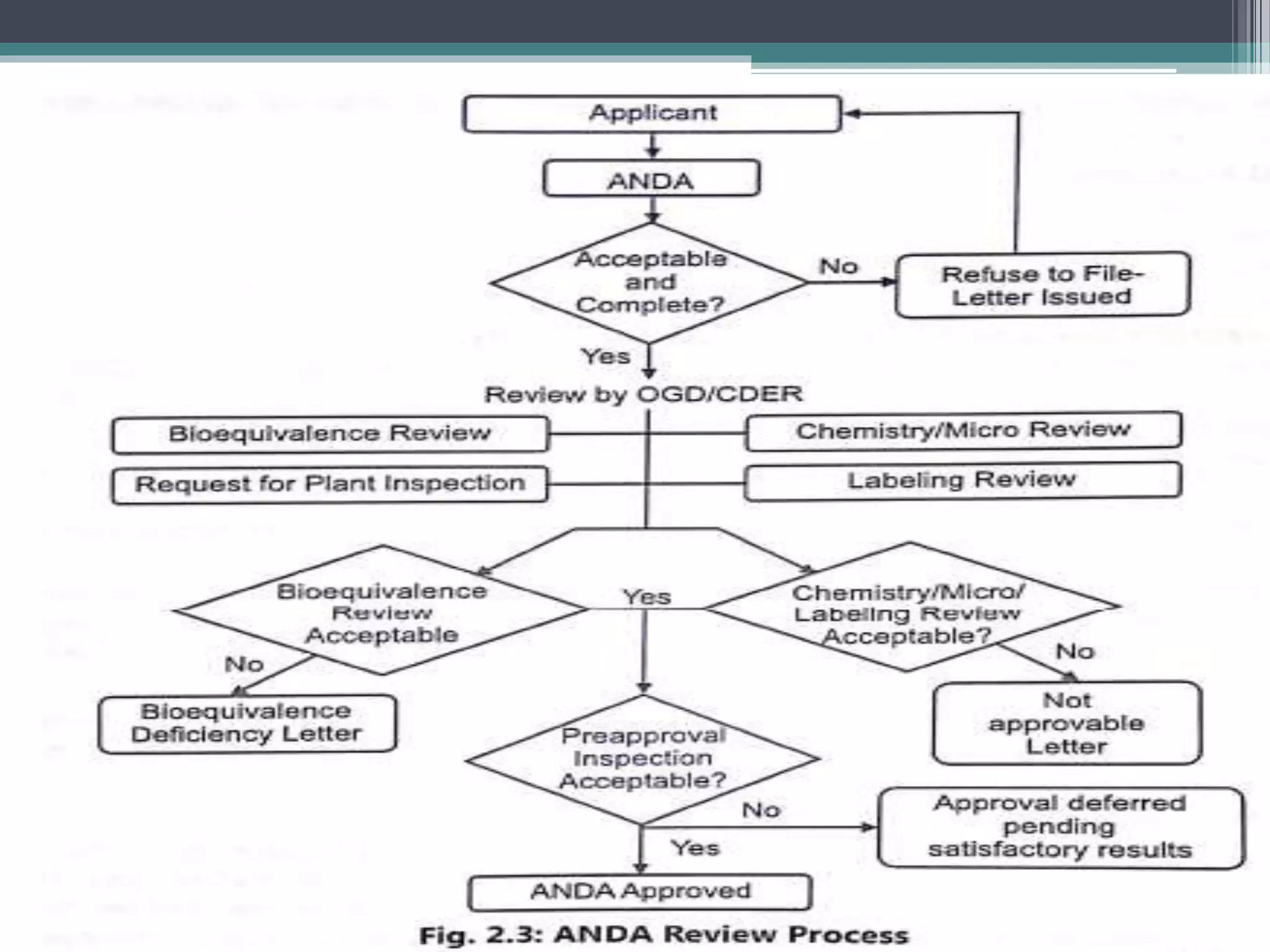
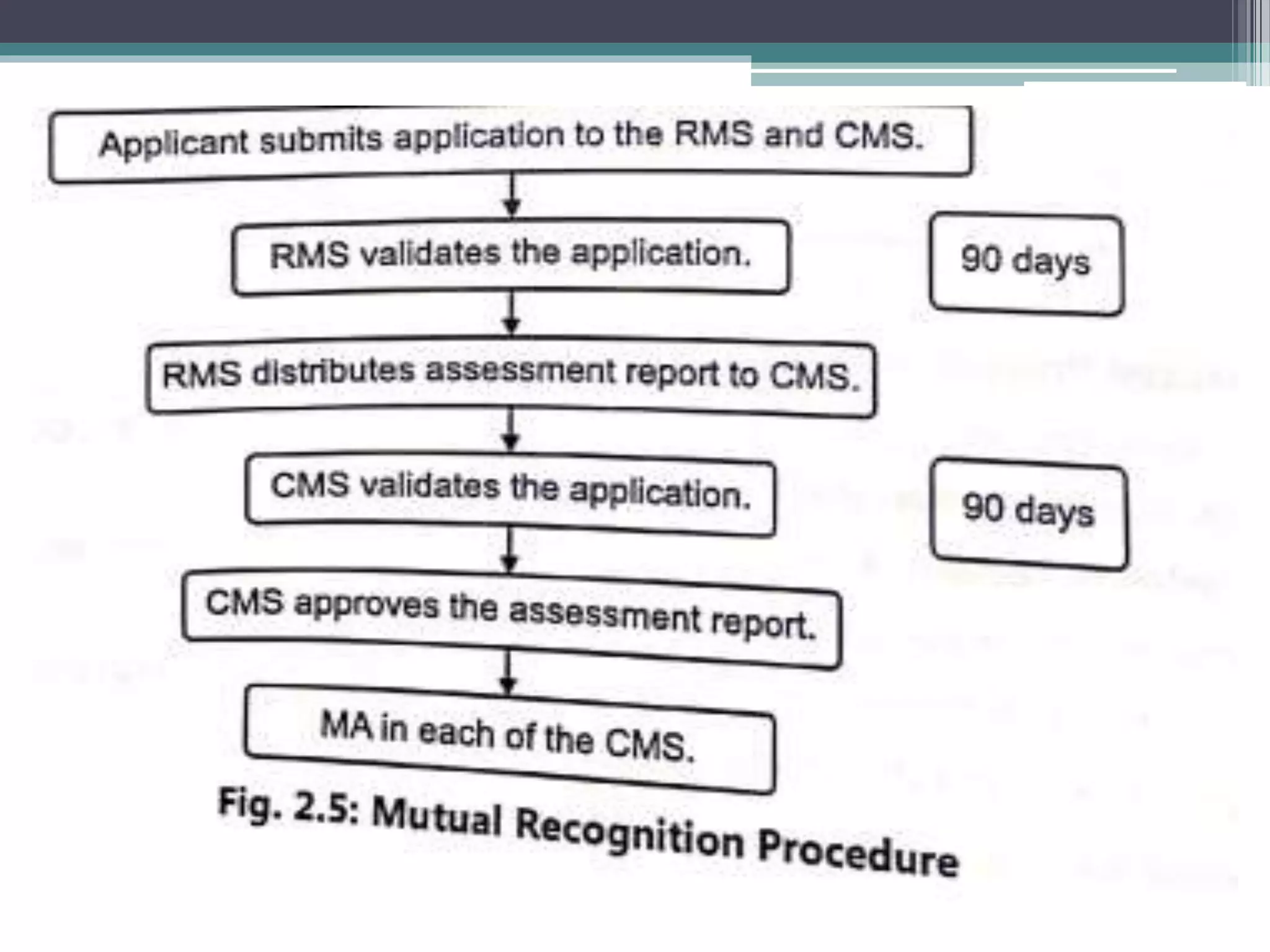
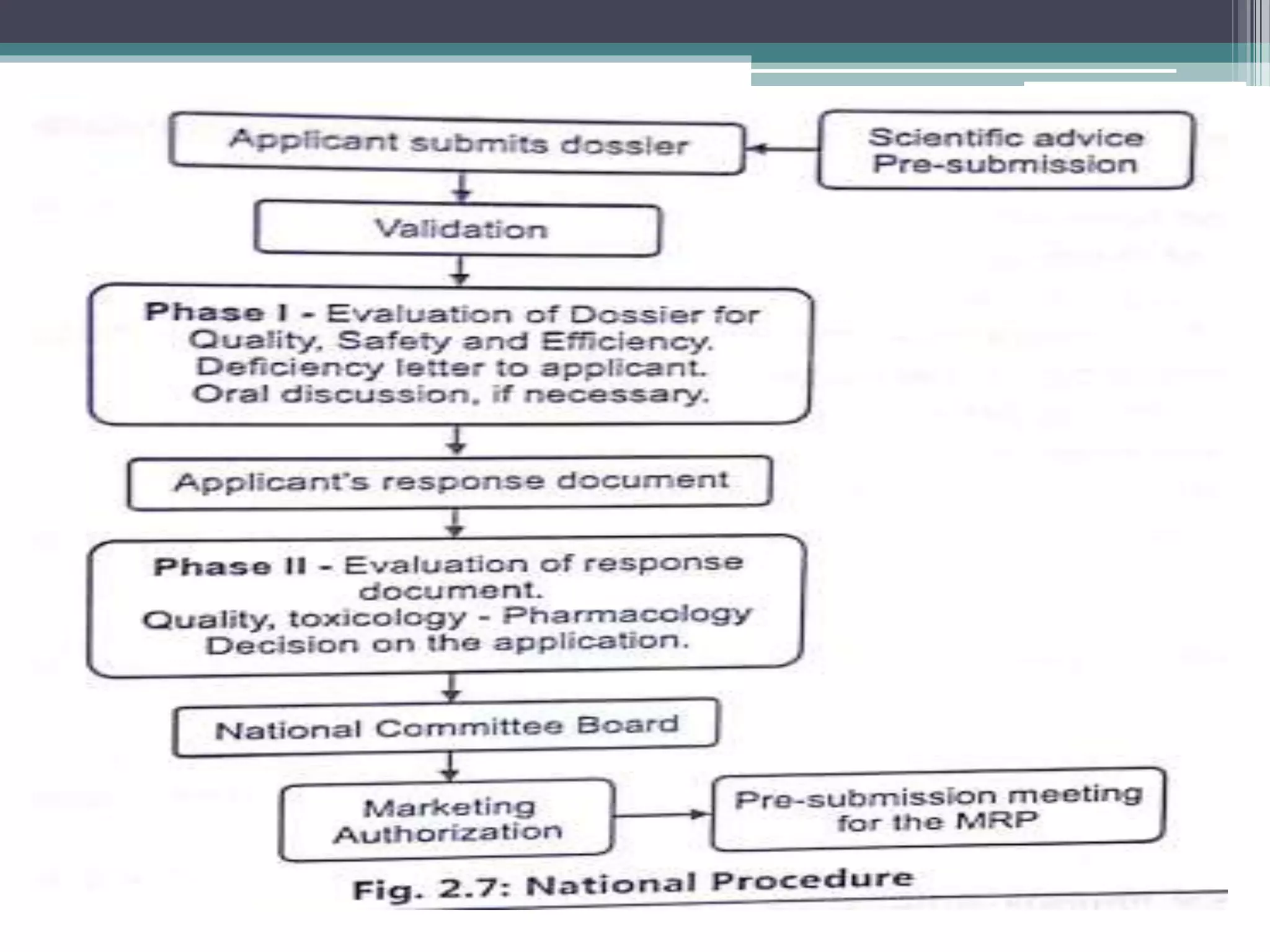
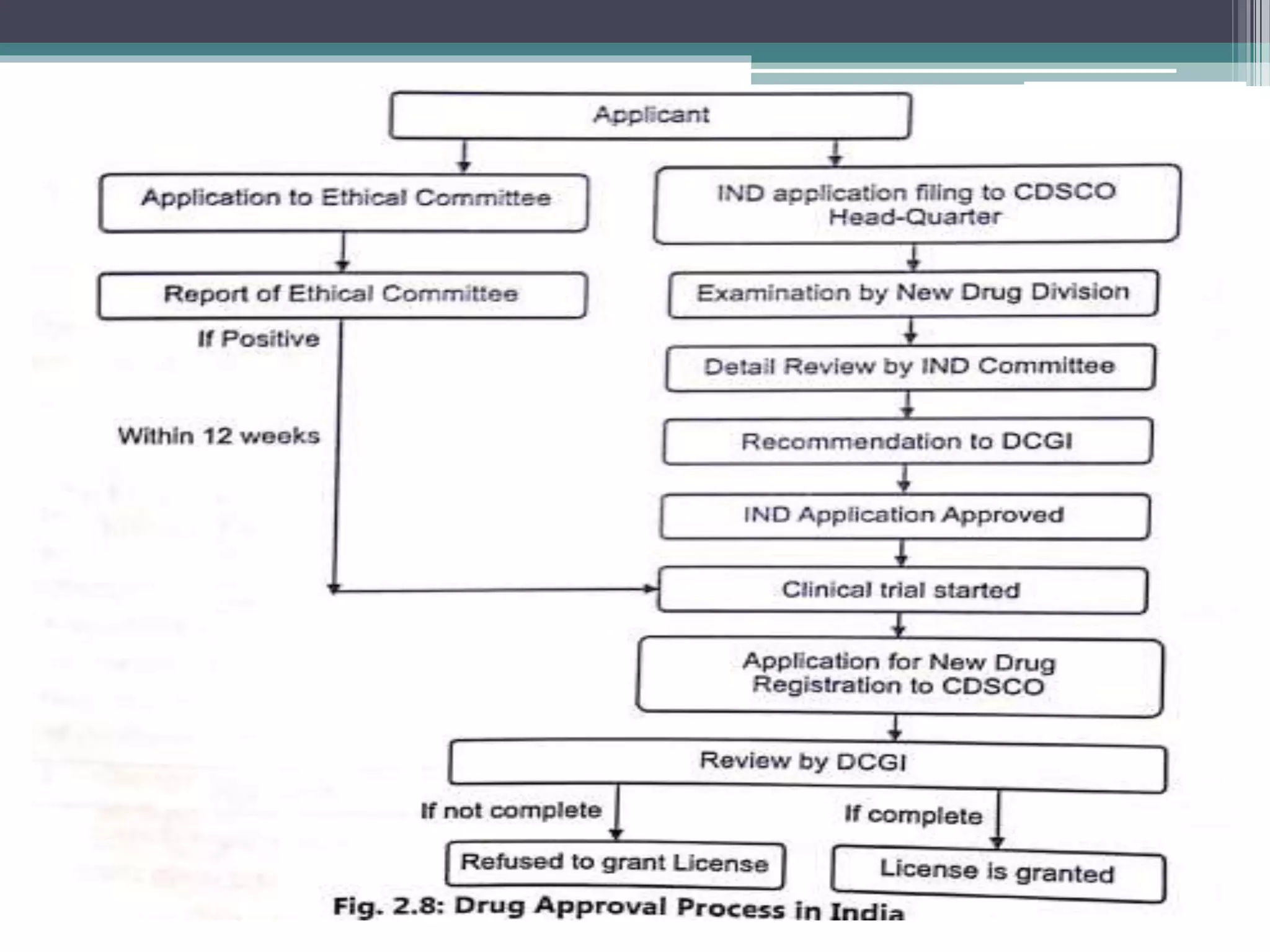
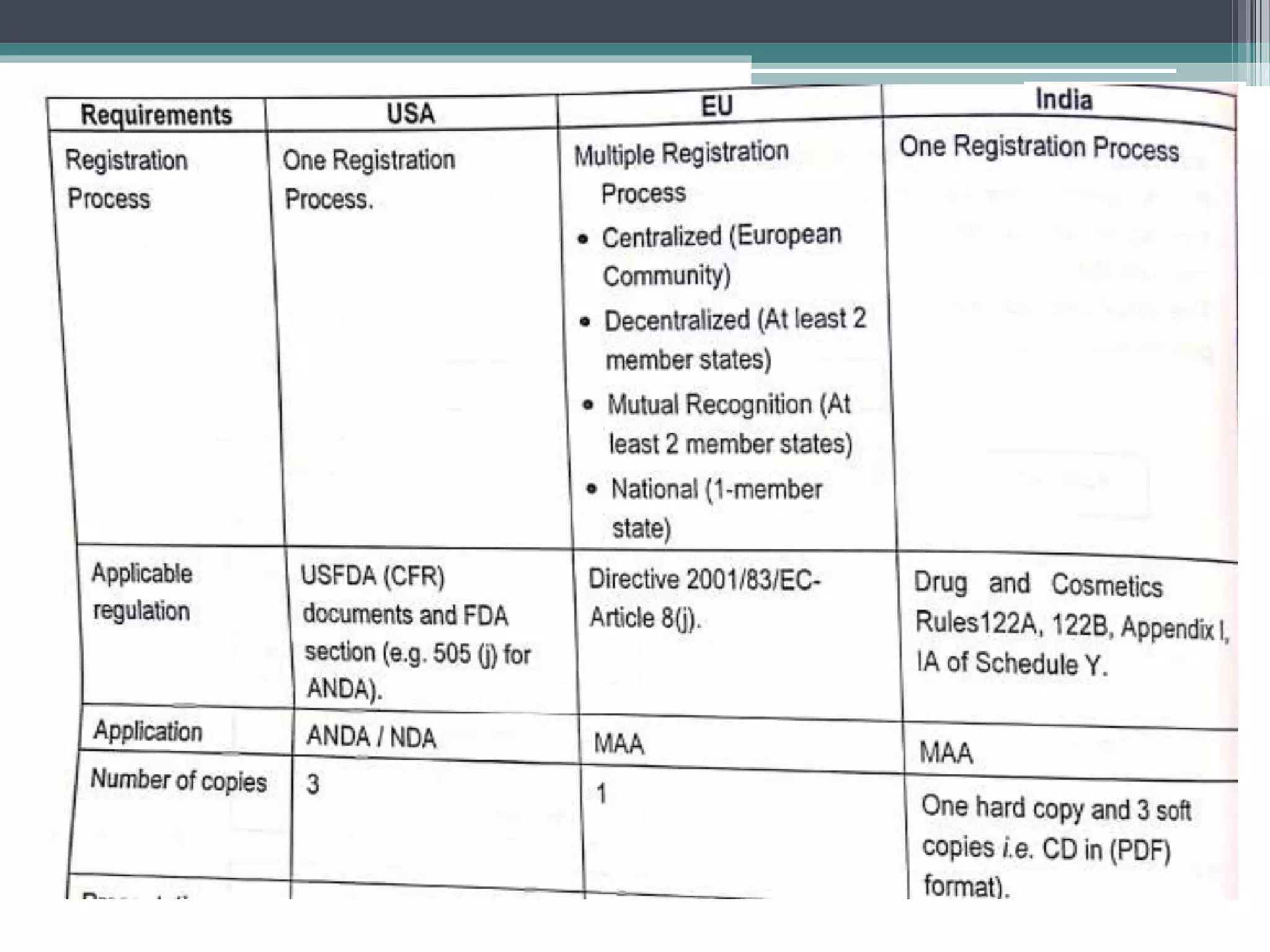
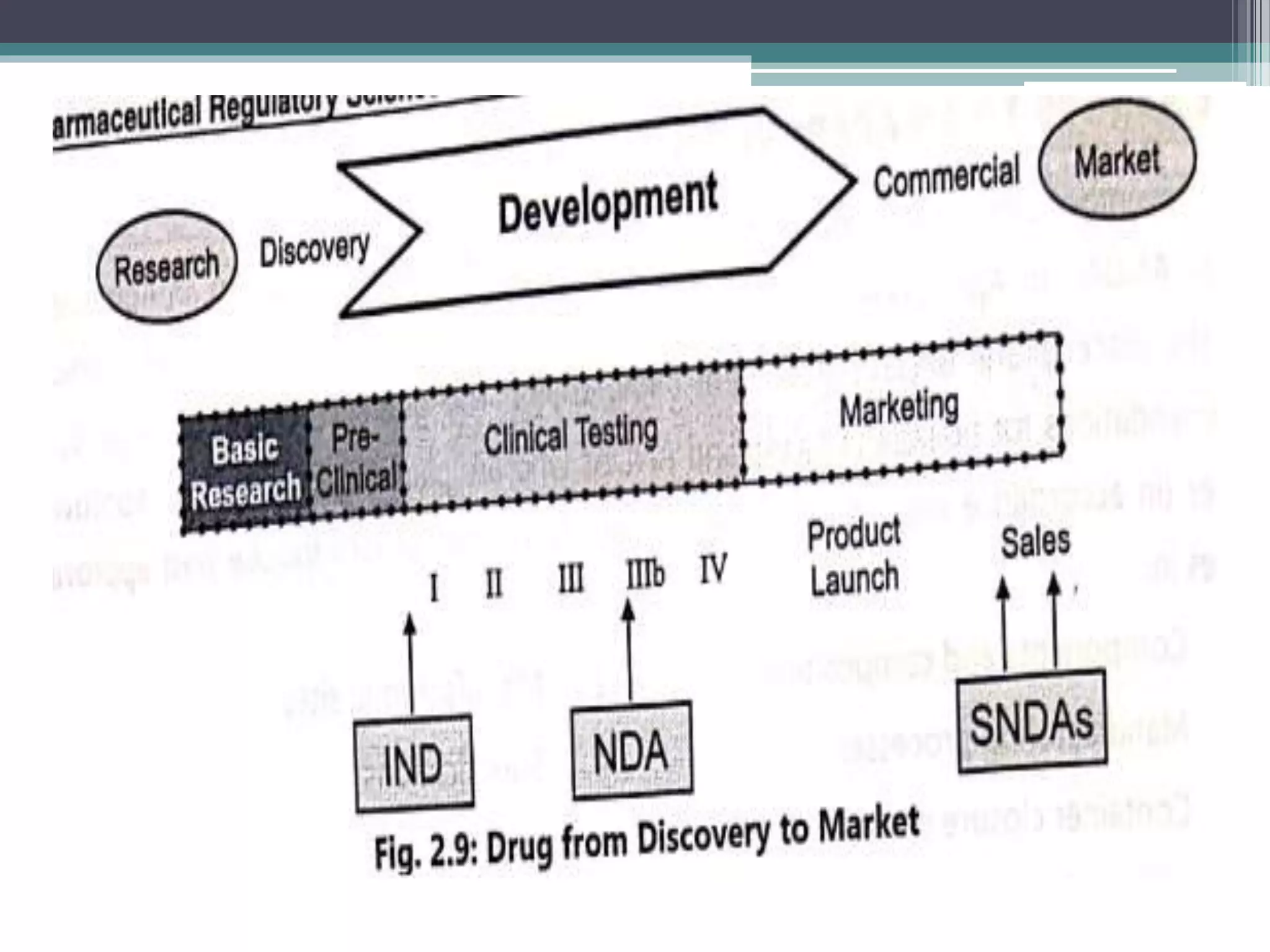
The document provides information on the regulatory approval process for drugs. It discusses the various stages of approval including investigational new drug applications (IND), new drug applications (NDA), and abbreviated new drug applications (ANDA).
The stages include pre-clinical testing, clinical trials through multiple phases, and regulatory review and approval. An IND must be approved by the FDA before clinical trials in humans can begin. If clinical trials are successful, manufacturers can file an NDA to request approval to market the drug. For generic drugs, an ANDA can be filed to demonstrate bioequivalence to an existing approved drug, without needing to re-conduct clinical trials. The approval process is complex and lengthy, usually taking 10-






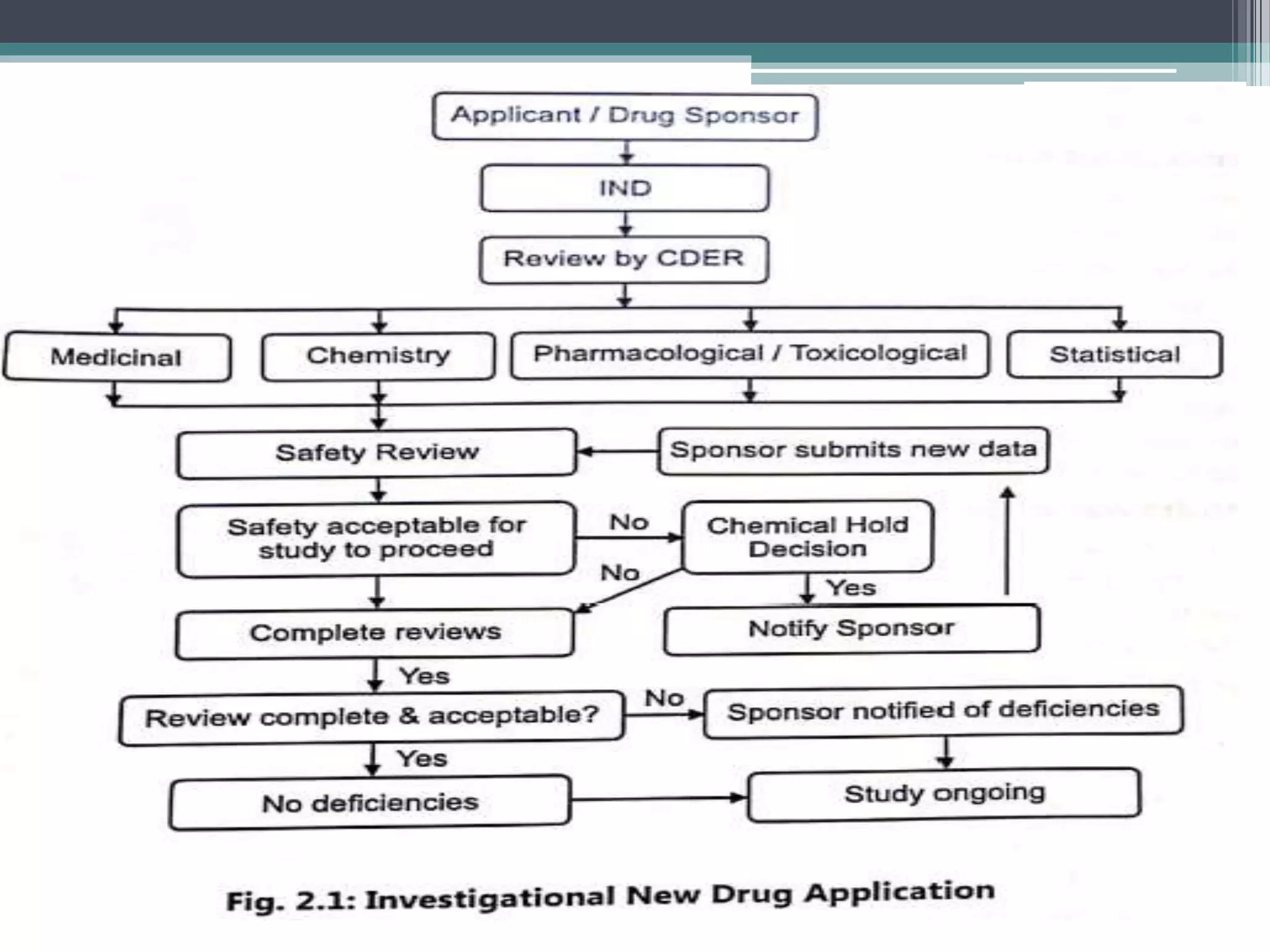



















































![Investigational New drug application [INDA]](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugapplicationinda-160619063044-thumbnail.jpg?width=640&height=640&fit=bounds)





![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)



































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)


