This document provides information on chronic myeloid leukemia (CML), including:
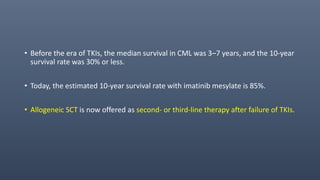
1. CML is driven by the BCR-ABL1 fusion gene resulting from the Philadelphia chromosome, and was historically treated with tyrosine kinase inhibitors (TKIs) like imatinib which improved 10-year survival to 85%.
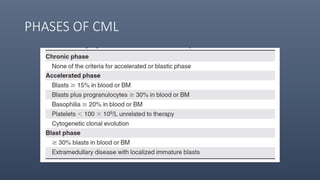
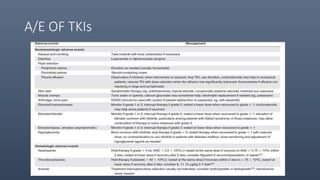
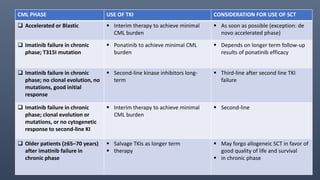
2. CML progresses through chronic, accelerated, and blast phases; TKIs can stabilize the disease in chronic phase and reduce transformation rates.
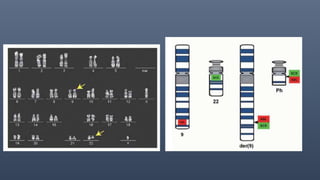
3. Diagnosis involves detecting the Philadelphia chromosome through cytogenetics of bone marrow samples and monitoring molecular response to TKIs is now standard practice.

































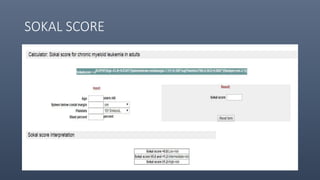
![• CML Prognostic models
• Sokal
• Hasford
• European Treatment and Outcome Study [EUTOS].
• Achievement of complete cytogenetic response has become the major therapeutic
endpoint and is the only endpoint associated with improvement in survival.
• The lack of achievement of major or complete molecular responses should not be
considered as “failure” of a particular TKI therapy and/or an indication to change the
TKI or to consider allogeneic SCT.](https://image.slidesharecdn.com/meloproliferativeneoplasms-1-190101165204/85/Meloproliferative-neoplasms-1-36-320.jpg)





































































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)

