Downloaded 54 times





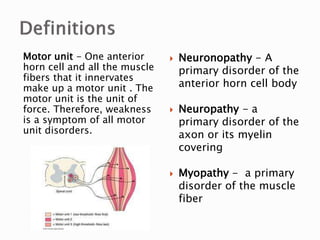







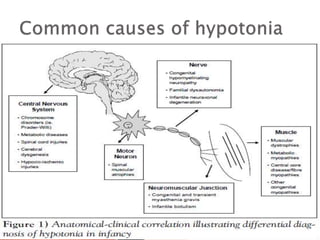







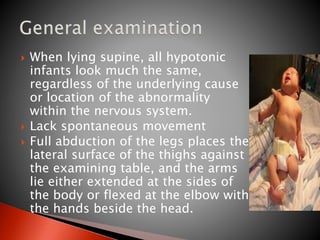
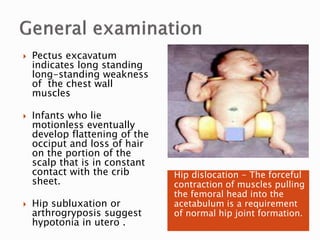





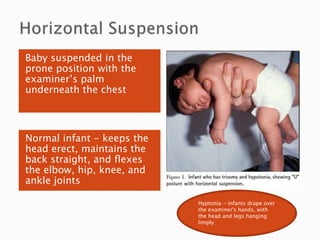






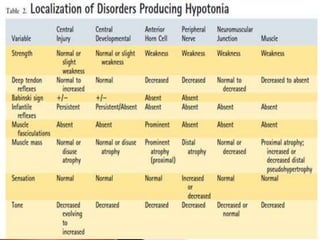
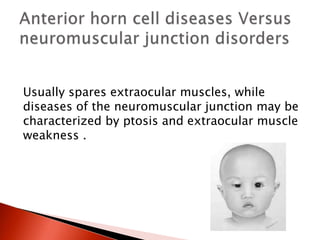




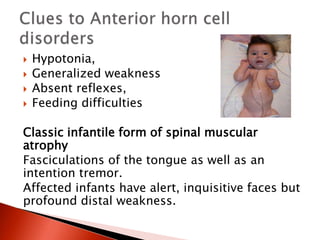





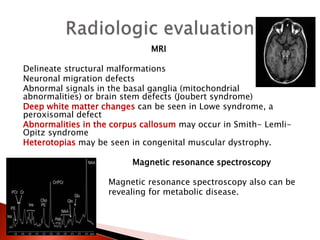


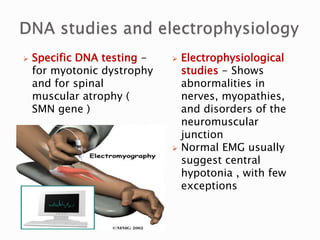


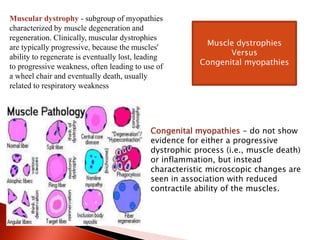

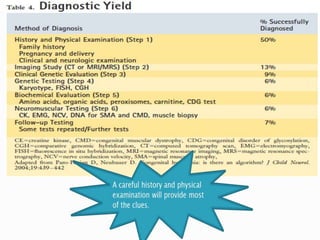
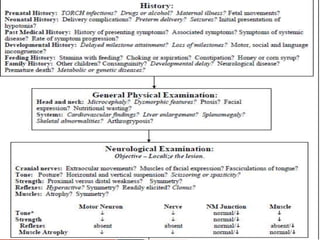



The document discusses hypotonia in infants and provides details on: - The differential diagnosis of hypotonia includes both benign and serious conditions. - Hypotonia can be caused by central nervous system issues or peripheral nervous system issues. Central causes account for 60-80% of cases. - The evaluation of an infant with hypotonia includes a detailed history, physical exam focusing on tone and strength, and initial screening tests. Further testing may include imaging, genetic testing, and metabolic testing depending on exam findings.





























































































