Downloaded 22 times
















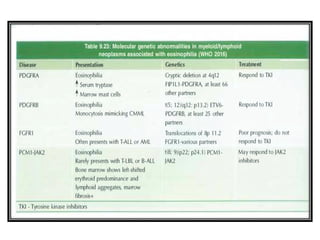




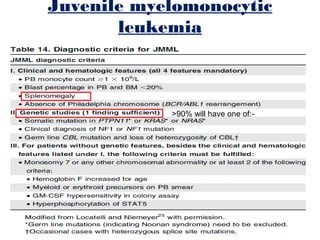


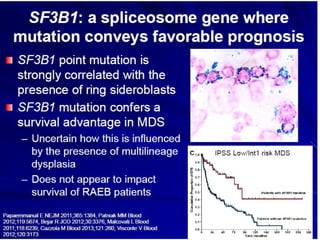

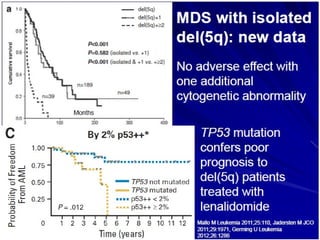





















The document summarizes recent changes to the World Health Organization's classification of myeloid neoplasms and acute leukemias based on advances since the 2008 classification. Key changes include new entities recognized based on unique biomarkers identified by gene expression analysis and sequencing. Entities were modified to better incorporate prognostic markers. Notable revisions include changes to criteria for chronic myeloid leukemia, myeloproliferative neoplasms, and myelodysplastic/myeloproliferative neoplasms. The classification of myelodysplastic syndromes was also updated to focus more on dysplasia levels than specific cytopenias. Recognition of myeloid neoplasms with germline predisposition was another major change.









































































