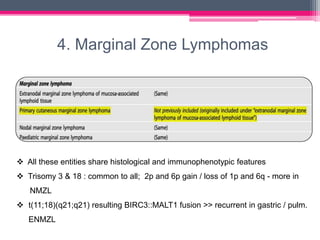
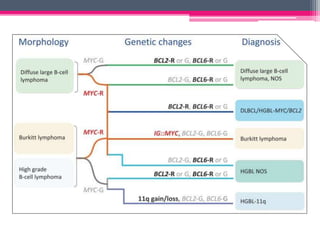
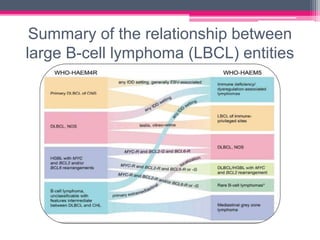
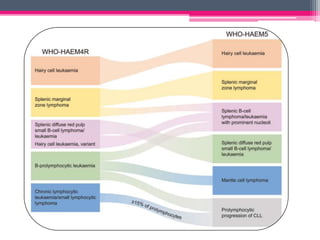
The WHO 2022 update on lymphoid malignancies made several changes to the classification of B cell neoplasms including recognizing new entities, refining existing entities, and standardizing nomenclature. Key changes included recognizing splenic B-cell lymphoma/leukaemia with prominent nucleoli as a distinct entity, reclassifying B-cell prolymphocytic leukaemia, updating the classification of Burkitt lymphoma to recognize EBV-positive and EBV-negative subtypes, and grouping diffuse large B-cell lymphomas into 17 specific entities or the category of high-grade B-cell lymphoma, NOS based on genetic and phenotypic features.













![3. Lymphoplasmacytic Lymphoma
• Two subtypes
IgM-LPL/ Waldenström Macroglobulinaemia (WM) type [most common]
Non-WM type LPL represents around 5% of LPL and includes:
-IgG or IgA monoclonal proteins
-Non-secretory LPL
-IgM LPL without BM involvement
• MYD88 mutation : hallmark driver mutation in the vast majority of LPL
(>90%) [To differentiate NMZL and ENMZL with plasmacytoid
differentiation and Multiple myeloma]
• Desirable to perform CXCR4 mutational analysis , esp. in patients planned
for Ibrutinib therapy](https://image.slidesharecdn.com/who5thhematolymphoidbcellneoplasm-230519003635-b1e4960f/85/who-5th-hematolymphoid-B-cell-neoplasm-pptx-17-320.jpg)