Downloaded 171 times

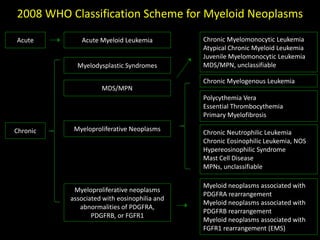





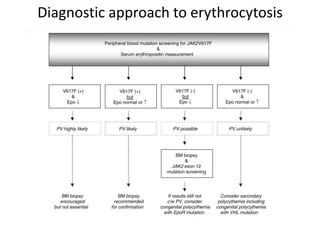




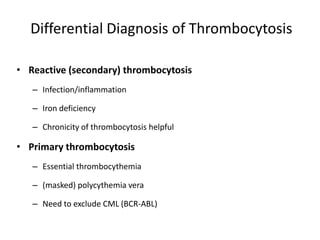


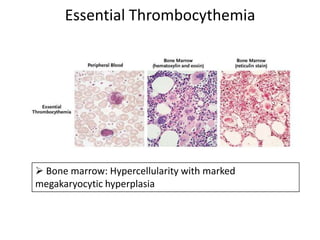

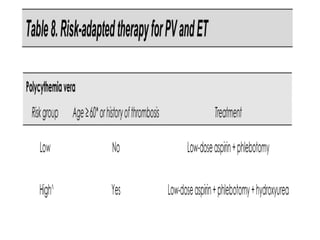
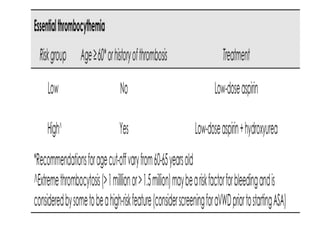




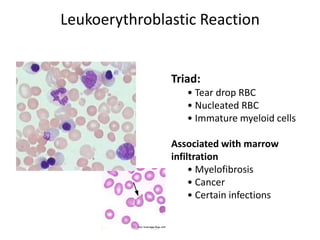

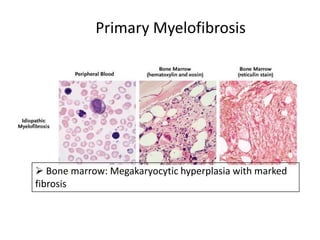


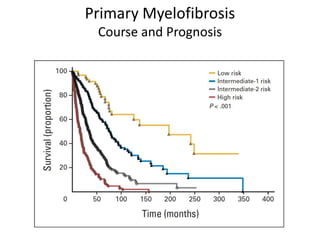


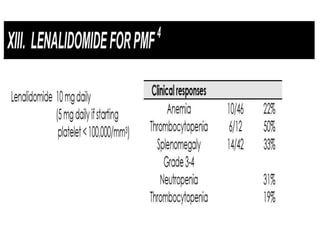
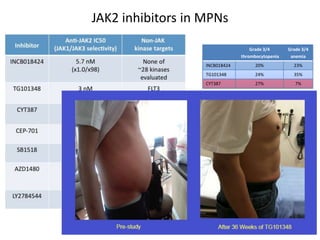

This document discusses the diagnostic approach and classification of myeloproliferative neoplasms according to the 2008 WHO criteria. It describes the differential diagnosis and clinical features of polycythemia vera, essential thrombocythemia, and primary myelofibrosis. It discusses diagnostic criteria, disease course, prognosis, and treatment approaches for these chronic myeloproliferative disorders.














































































