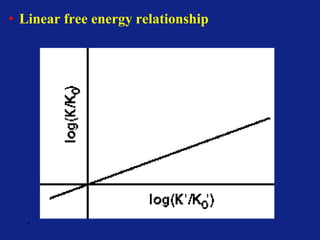
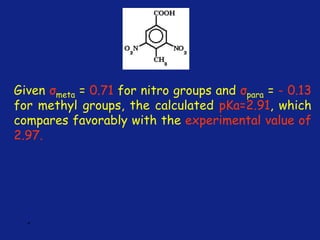
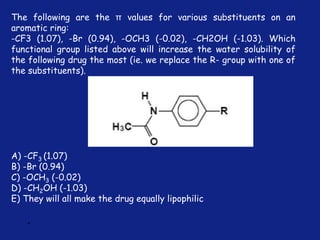
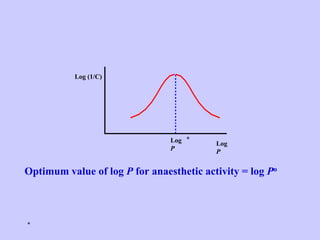
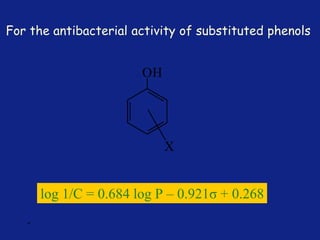
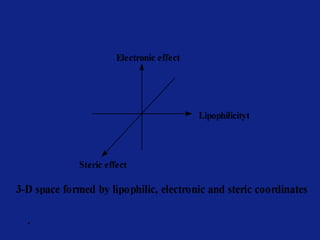
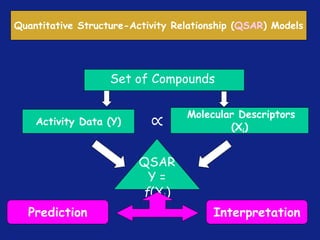
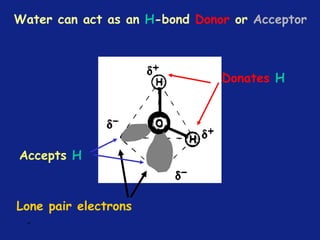
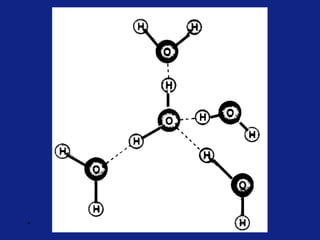
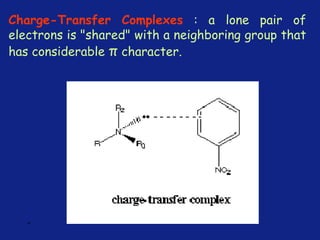
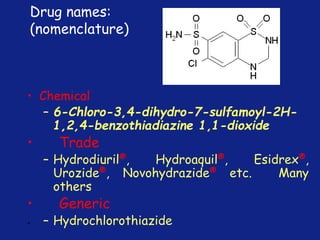
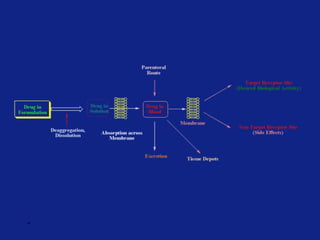
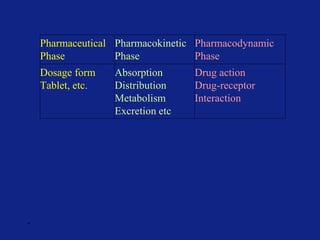
Medicinal chemistry involves the design and discovery of biologically active compounds to treat diseases. A drug's effects depend on its physicochemical properties like ionization, polarity, and lipid-water partition coefficient, which determine absorption, distribution, metabolism, and excretion. Drug names include chemical, trade, and generic names. Understanding a drug's physicochemical parameters helps explain its mechanism of action and effects.















![*
Electronic Effects
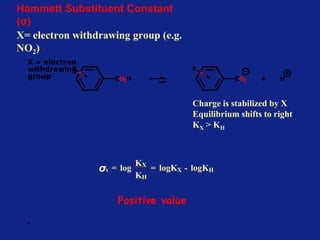
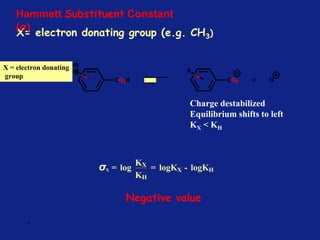
Hammett Substituent Constant
(σ)
• The constant (σ) a measure of the e-withdrawing or e-
donating influence of substituents
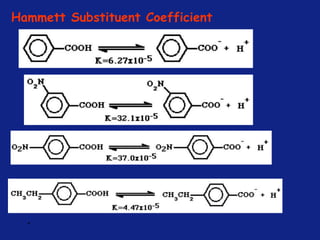
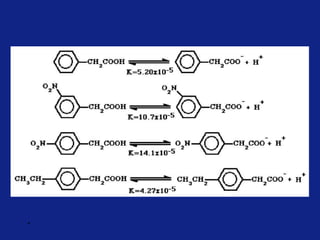
• It can be measured experimentally and tabulated
(e.g. s for aromatic substituents is measured by comparing the
dissociation constants of substituted benzoic acids with benzoic
acid)
X=H KH = Dissociation constant= [PhCO2-]
[PhCO2H
]](https://image.slidesharecdn.com/medchemunit2-181128103027/85/Med-chem-unit2-18-320.jpg)