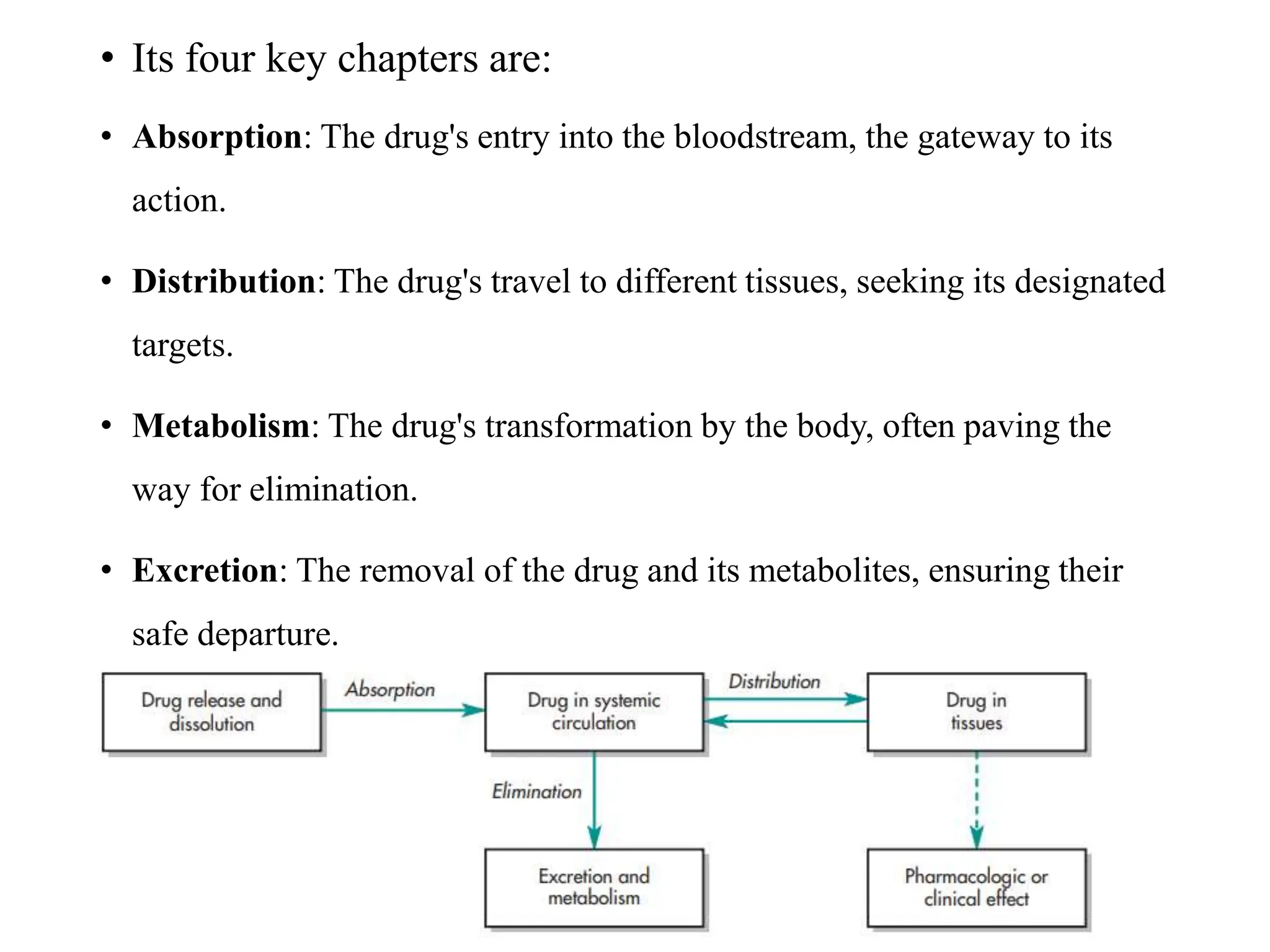
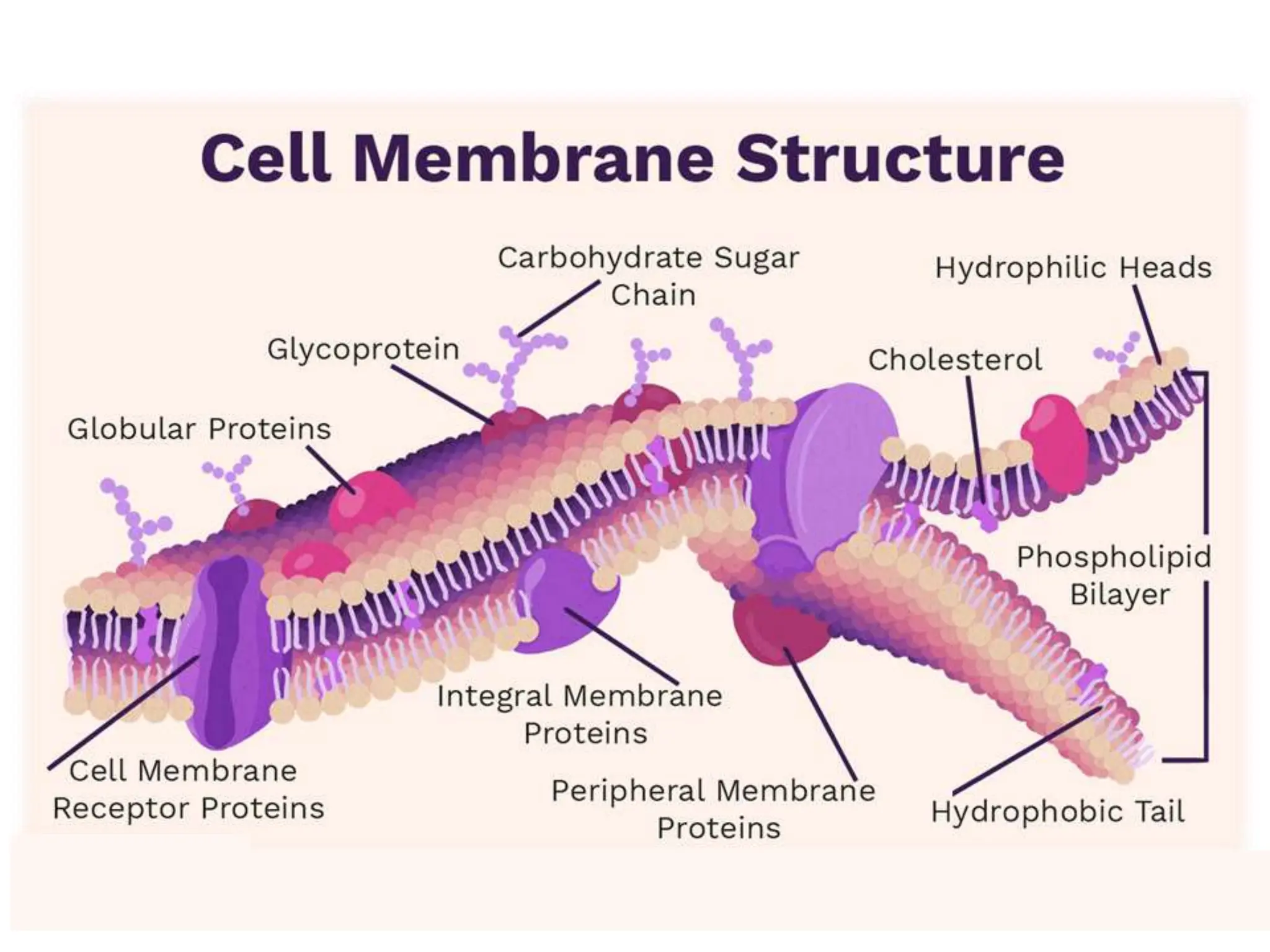
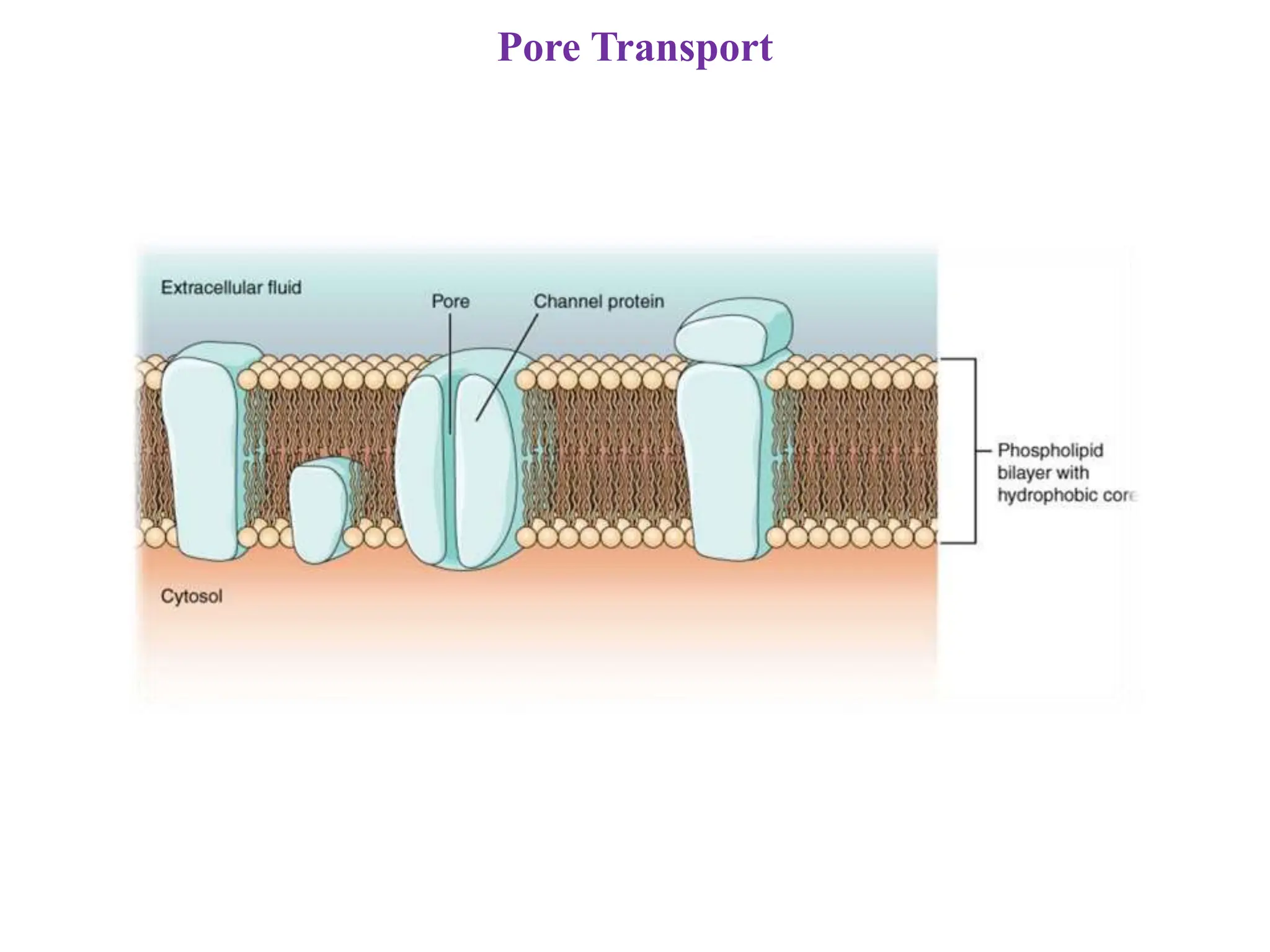
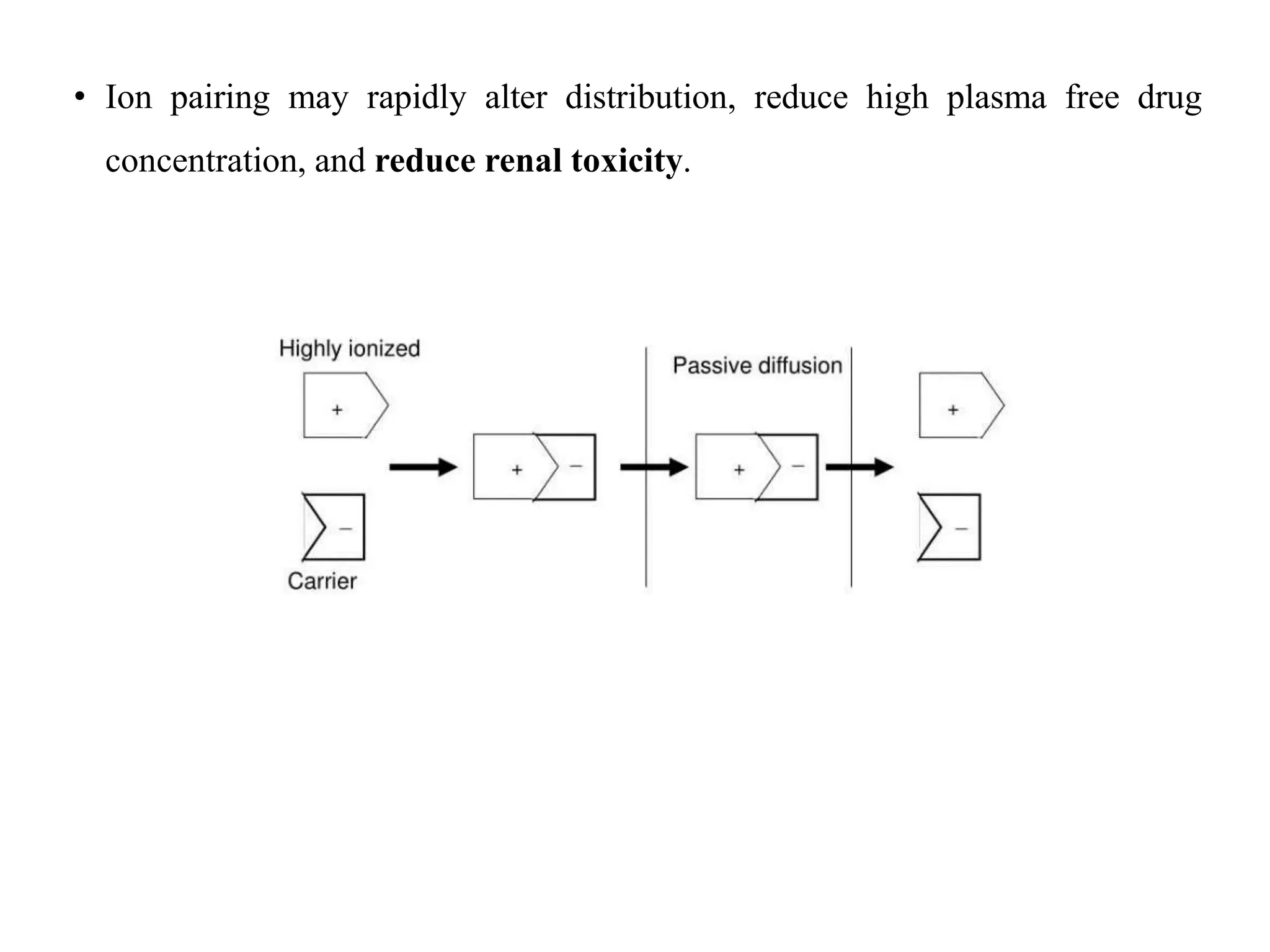
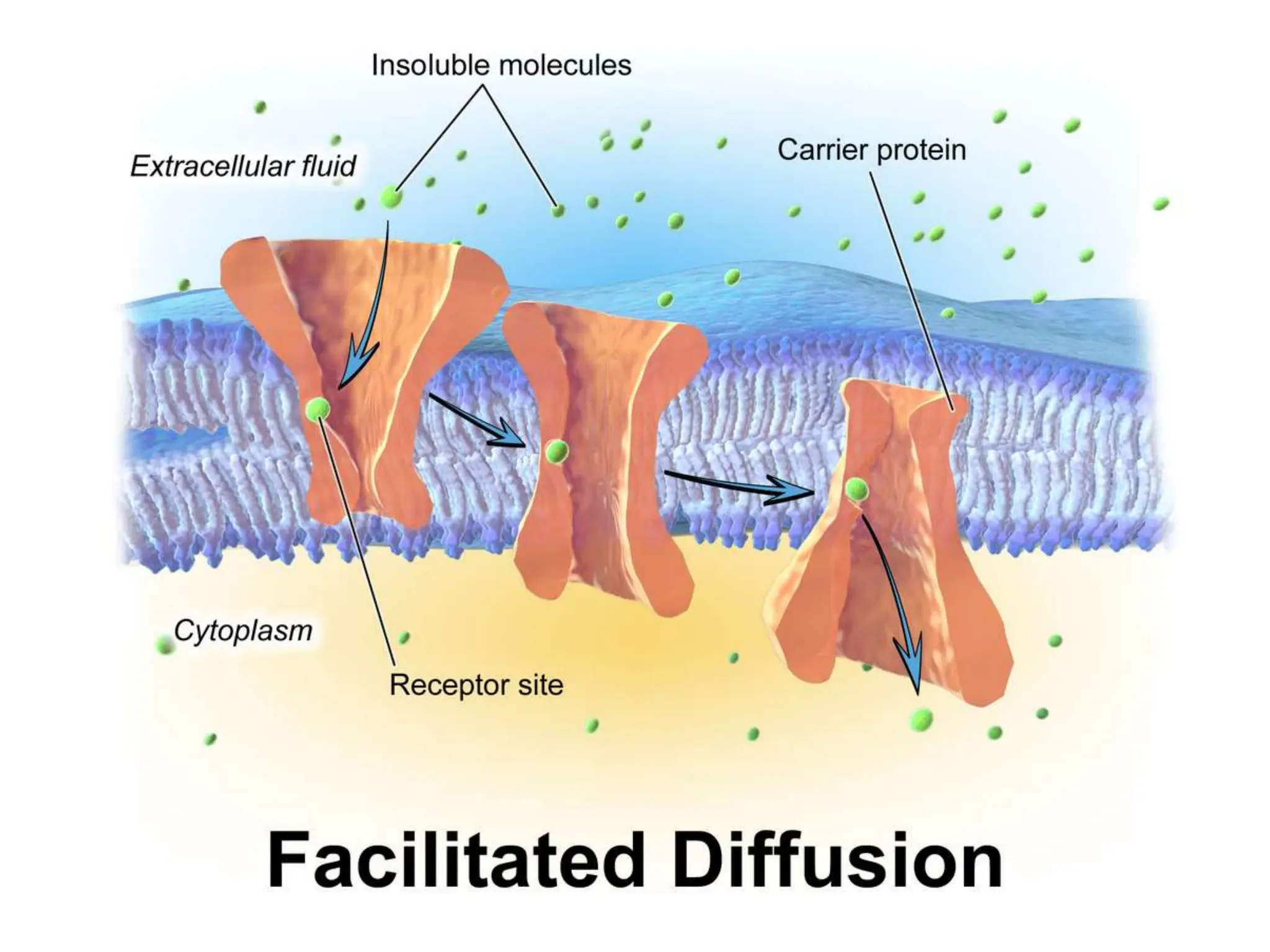
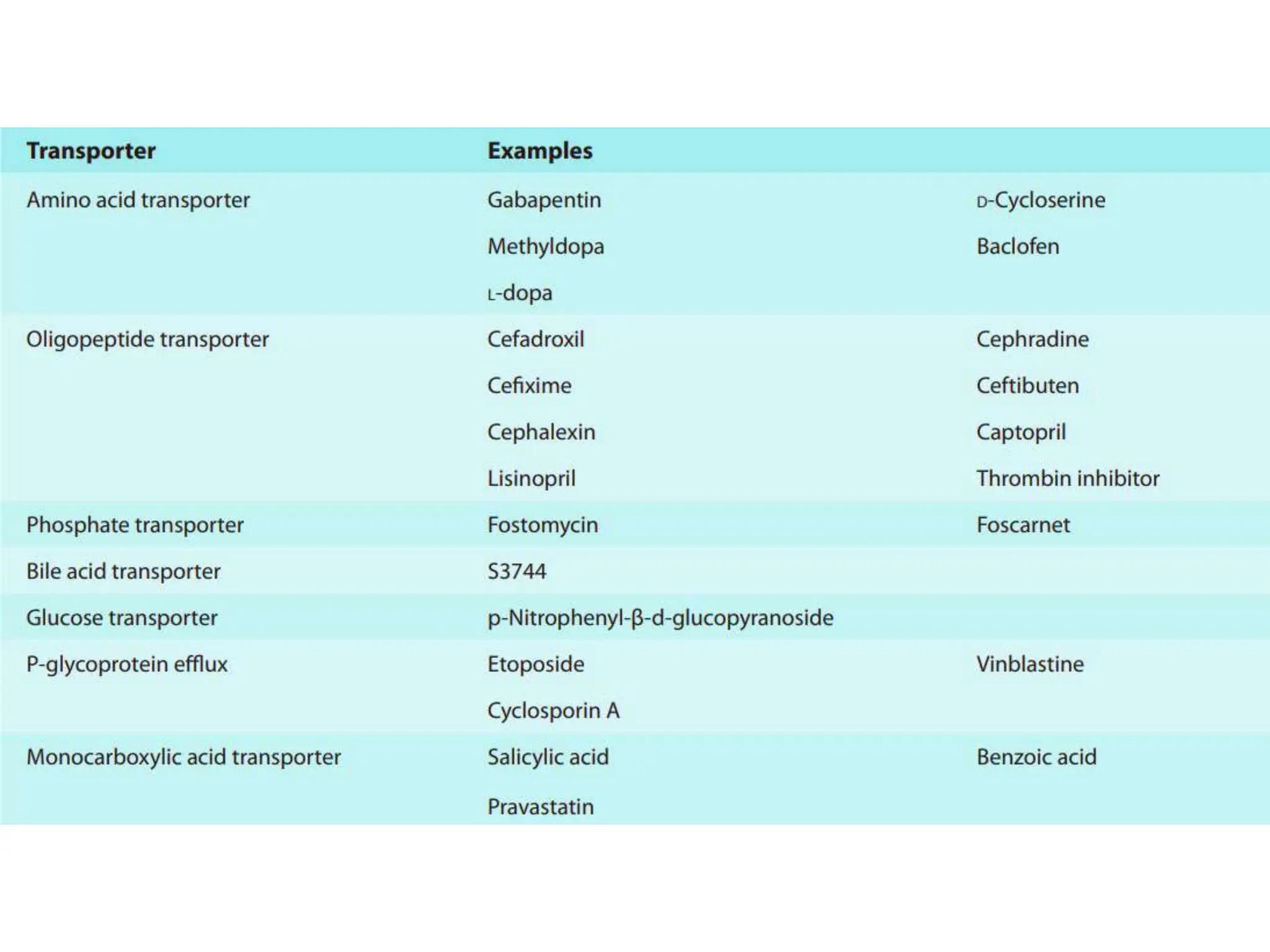
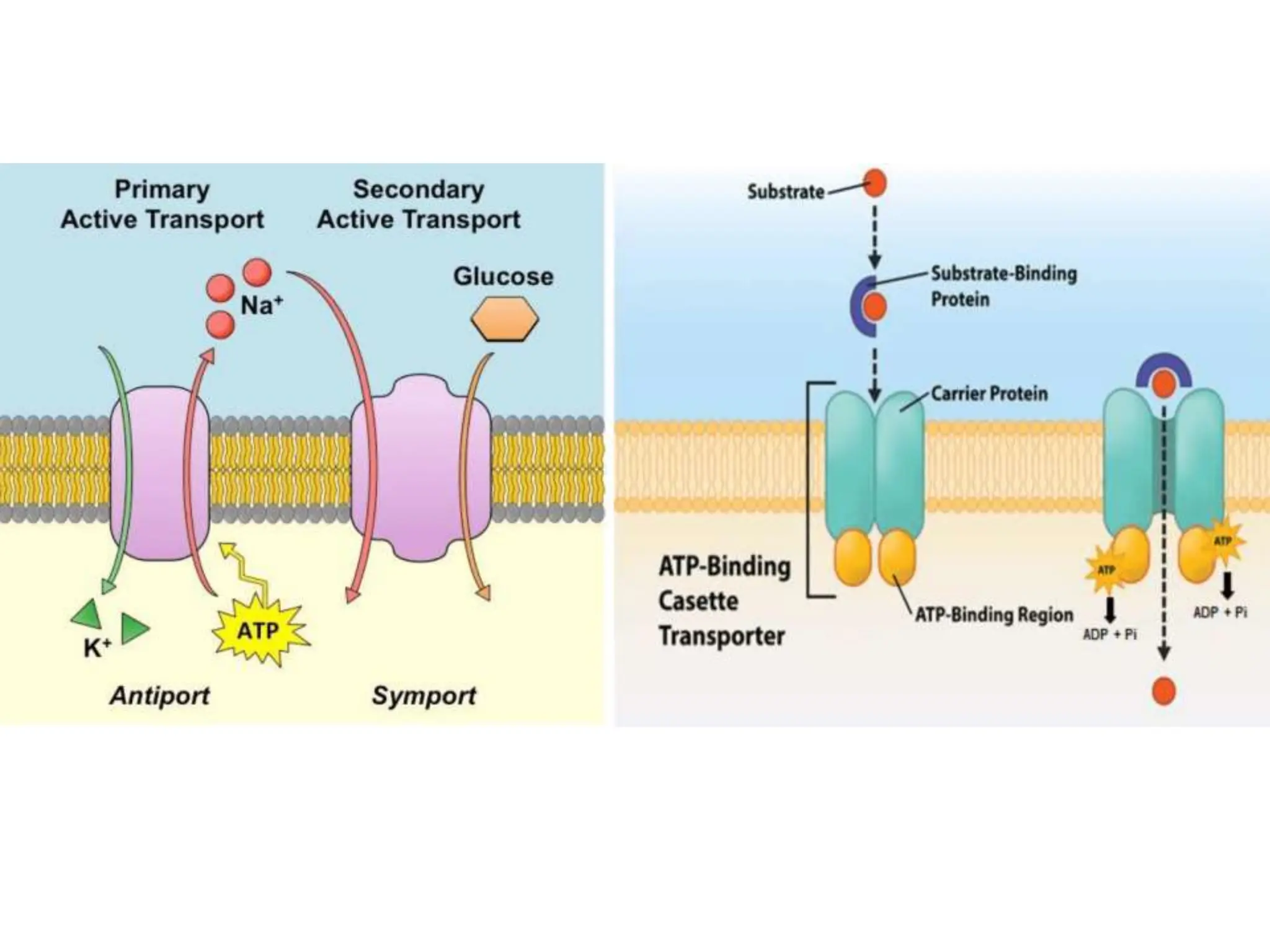
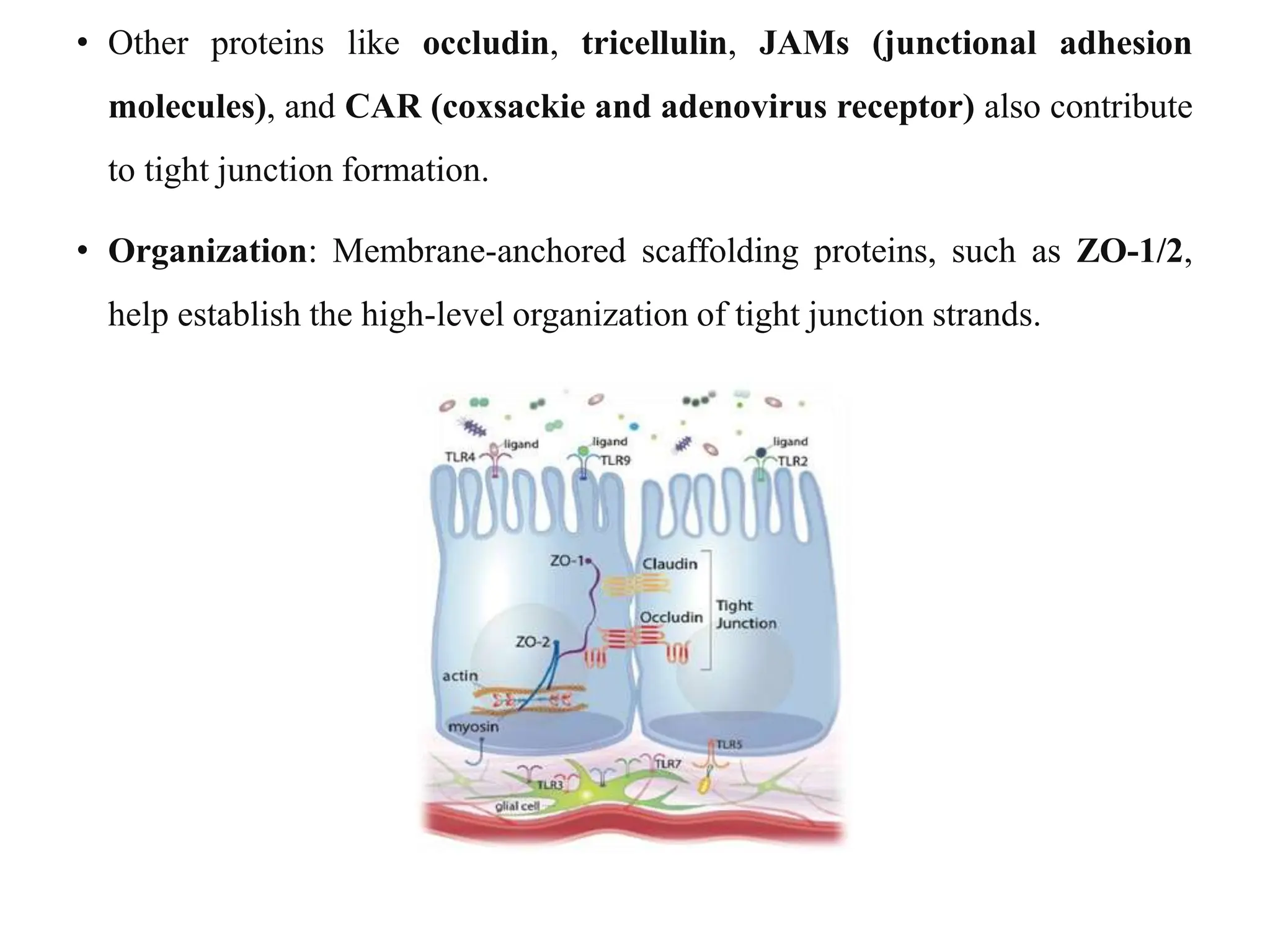
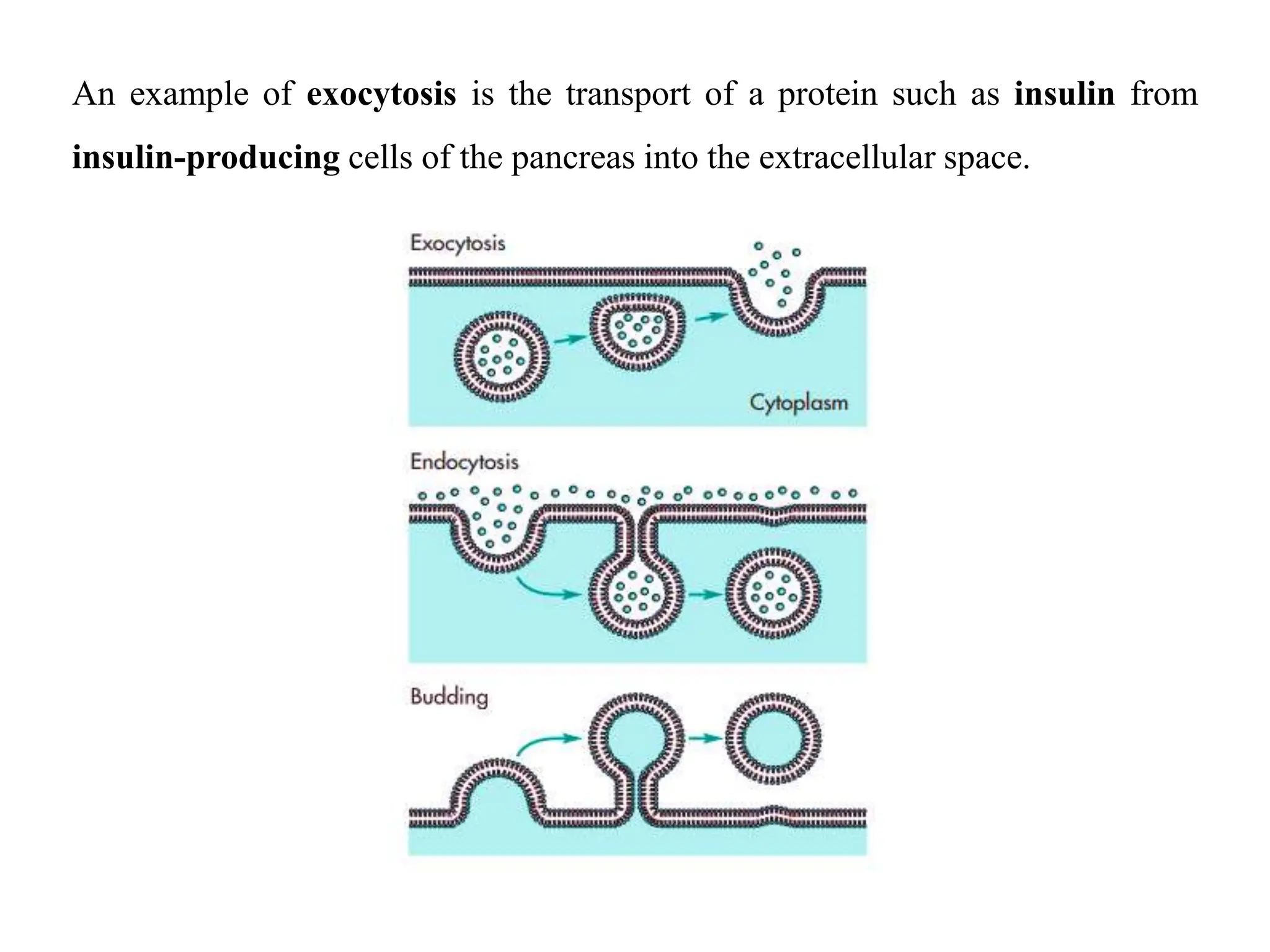
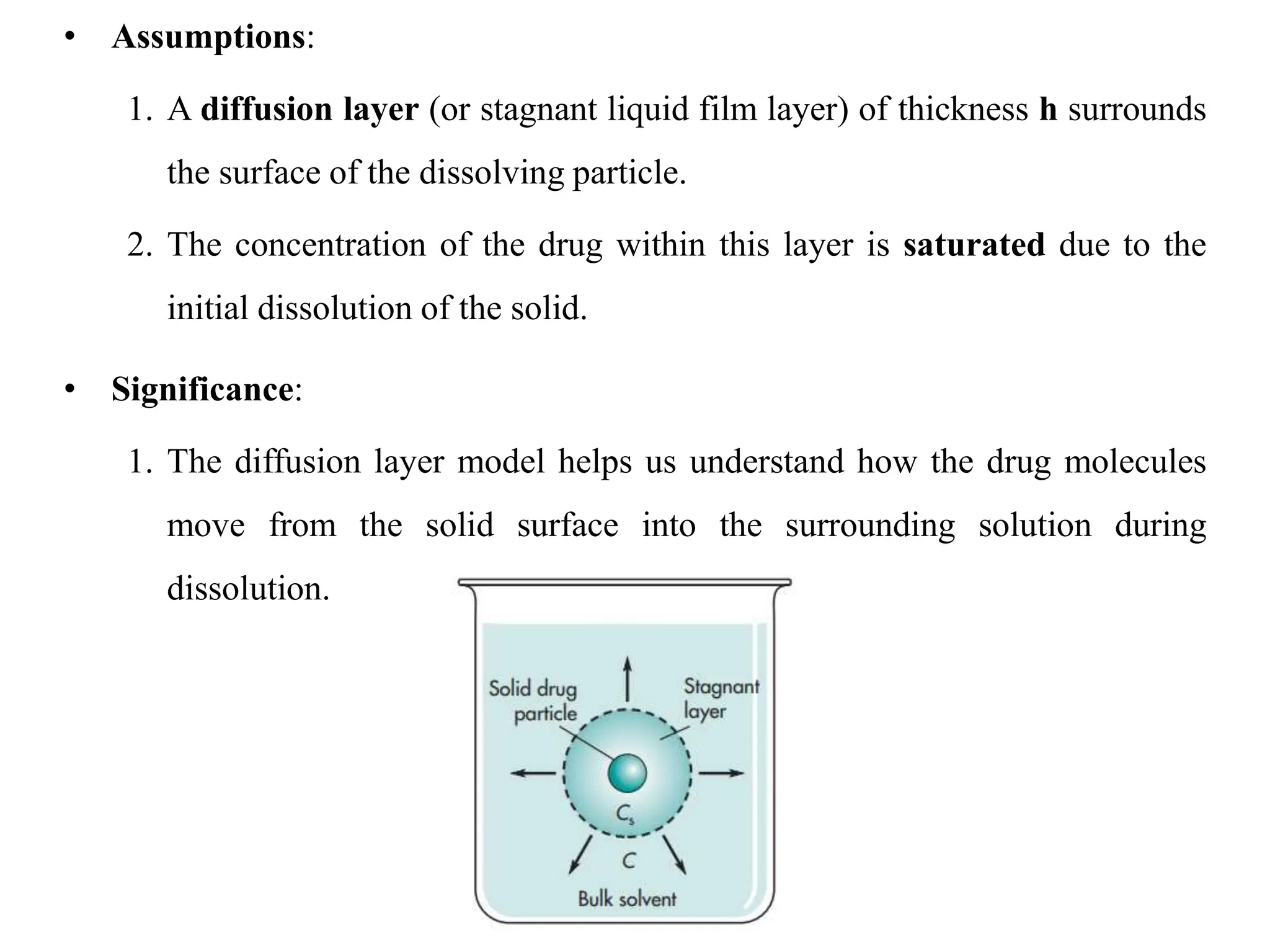
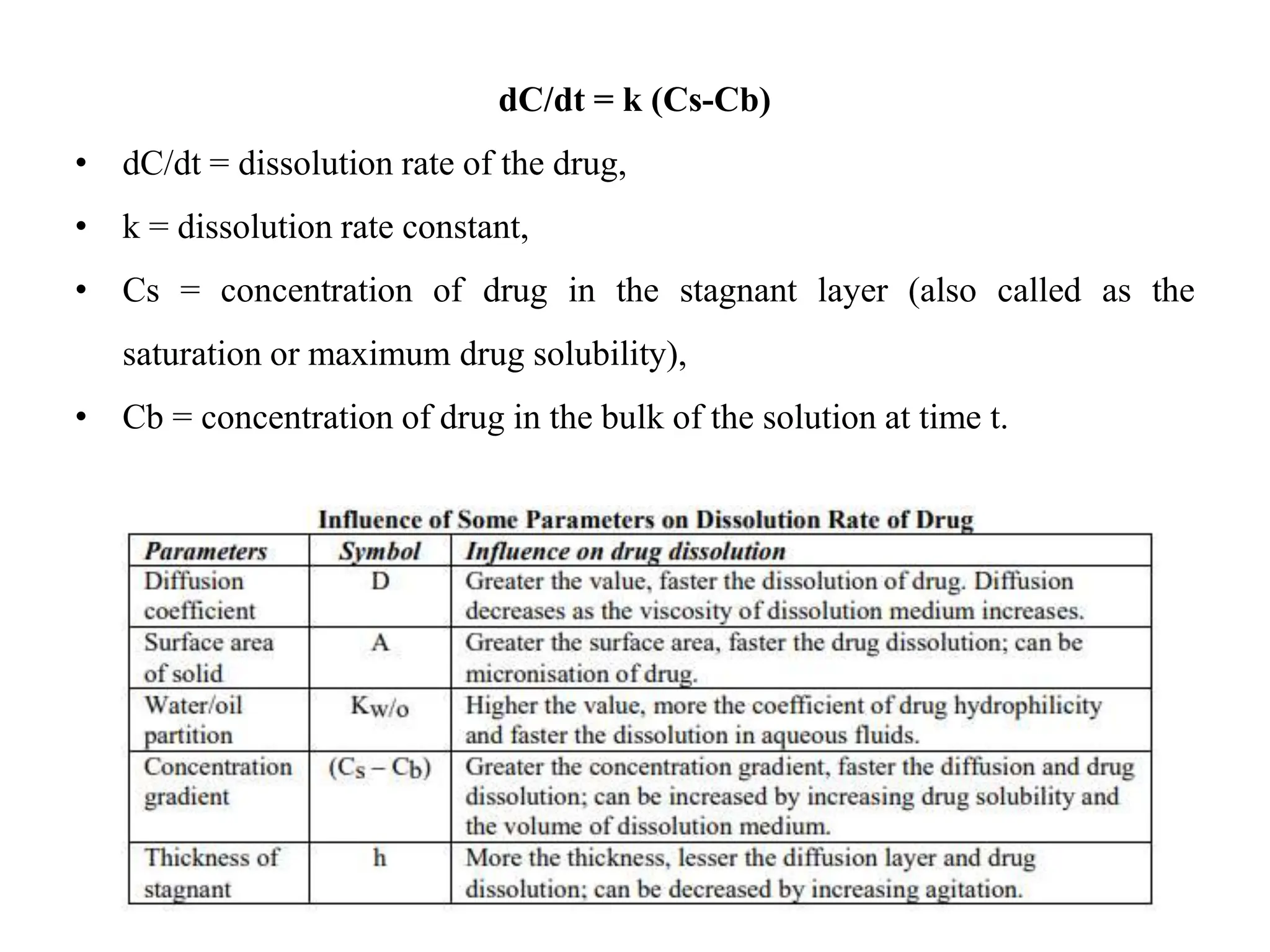
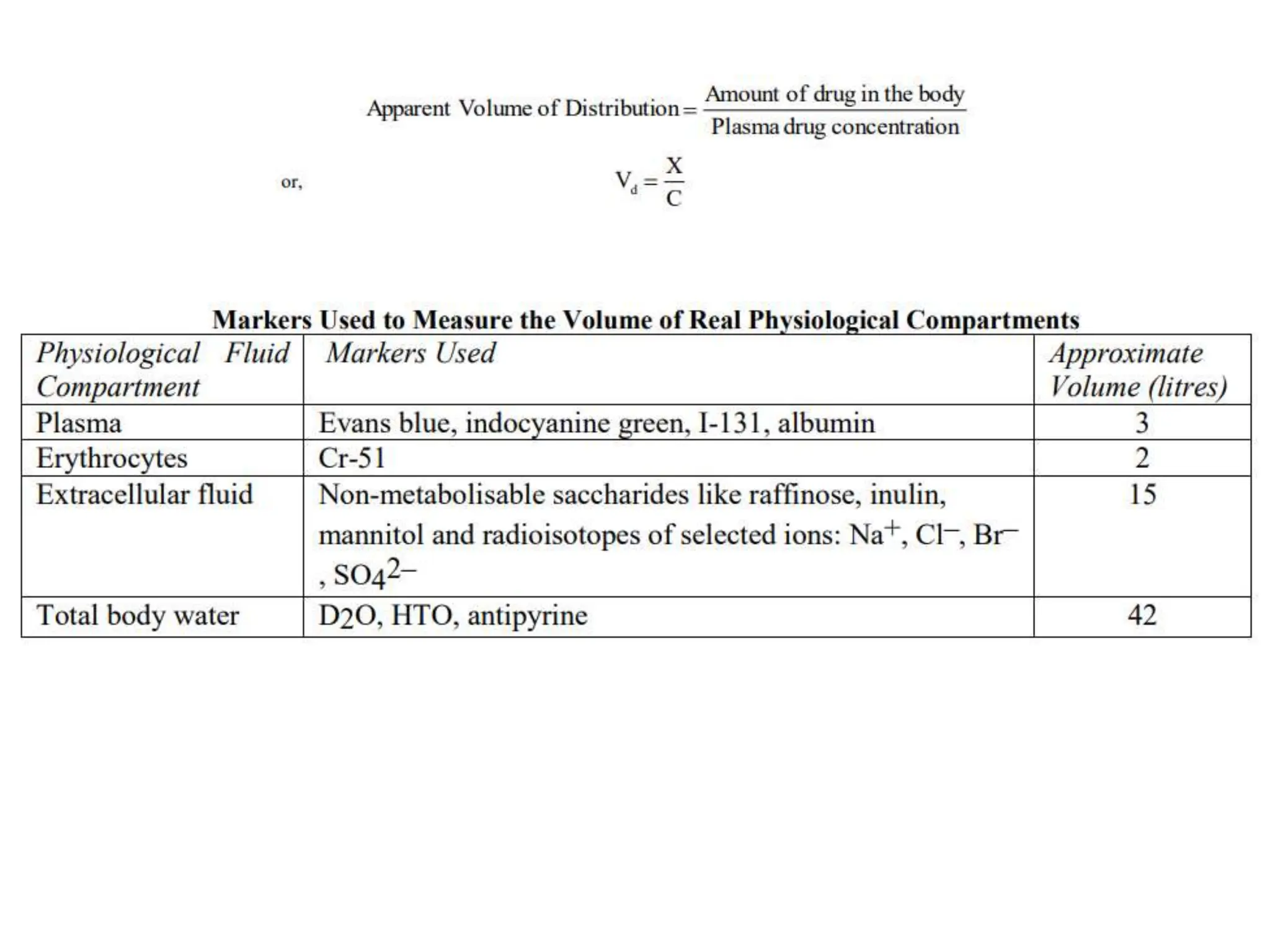
This document provides an introduction to biopharmaceutics and pharmacokinetics. It discusses how biopharmaceutics examines the relationship between a drug's physical/chemical properties, dosage form, and route of administration on systemic absorption. The four key aspects of pharmacokinetics are also introduced: absorption, distribution, metabolism, and excretion. Various mechanisms of drug absorption across cell membranes are then described in detail, including passive diffusion, active transport, and carrier-mediated transport systems.