











![pKa Determination
The solubility and absorption can be altered with changing pH.
pKa is dissociation constant of drug.
Unionized drug is lipid soluble that is permeate through lipid membrane. But ionized
drug is lipid insoluble, slow permeation.
Acidic substance become ionized by [H+] release and basic substance become
unionized by accept [H+].
Weak acid become ionized in alkaline environment. Weak base become ionized in
acidic environment.
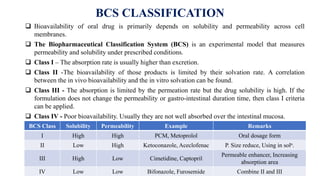
Henderson-Hasselbalch equation:
For base: pH = pKa + log
[𝑖𝑜𝑛𝑖𝑧𝑒𝑑]
[𝑢𝑛𝑖𝑜𝑛𝑖𝑧𝑒𝑑]
For acid: pH = pKa + log
[𝑢𝑛𝑖𝑜𝑛𝑖𝑧𝑒𝑑]
[𝑖𝑜𝑛𝑖𝑧𝑒𝑑]
% ionized =
10pH−pKa
1+10pK−pHaX100](https://image.slidesharecdn.com/unit-1preformulation-231103053135-1d5f3fb7/85/Unit-1-Preformulation-pptx-18-320.jpg)











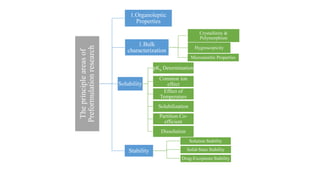
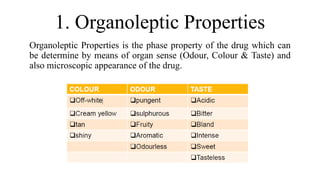
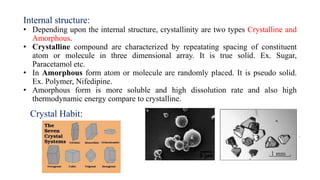
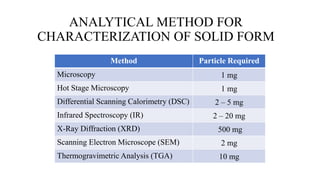
This document discusses preformulation studies, which characterize the physicochemical properties of new drug molecules to develop safe, effective, and stable dosage forms. It covers various areas of preformulation research like organoleptic properties, bulk characterization, crystallinity, polymorphism, hygroscopicity, micromeritic properties, solubility, pKa determination, and stability studies. Analytical techniques used for characterization include microscopy, DSC, IR, XRD, SEM, and TGA. The goals of preformulation are to establish the drug's properties, determine its kinetics and stability, ensure compatibility with excipients, and improve the drug product's manufacturing, storage and performance.







![Tablets •|• [types of tablets] and classification of tablets](https://cdn.slidesharecdn.com/ss_thumbnails/ip2-241009012105-21394589-thumbnail.jpg?width=640&height=640&fit=bounds)



























































