Downloaded 219 times





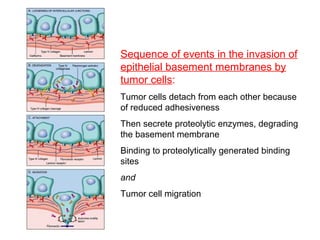



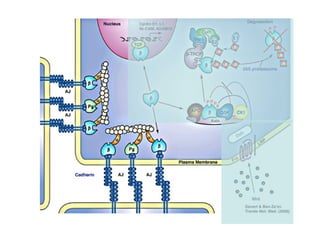


![2-Local degradation of the basement
membrane and interstitial connective tissue
Elaboration of proteases by
– Tumor cells themselves or
– Stromal cells [induced by tumor cells]
Many different families of proteases
– Matrix metalloproteinases (MMPs)
– Cathepsin D &
– Urokinase plasminogen activator](https://image.slidesharecdn.com/invasionmetastasis-csbrp-161201085229/85/Invasion-metastasis-csbrp-21-320.jpg)


![3 - Attachment to novel ECM components
• Normal epithelial cells have receptors, such as integrins,
for basement membrane laminin and collagens that are
polarized at their basal surface
– These receptors help to maintain the cells in a resting,
differentiated state
• Loss of adhesion in normal cells leads to induction of
apoptosis [tumor cells are resistant to this form of cell
death]
• The matrix itself is modified in ways that promote
invasion and metastasis
– Eg: cleavage of the basement membrane proteins collagen IV
and laminin by MMP2 or MMP9 generates novel sites that bind
to receptors on tumor cells and stimulate migration](https://image.slidesharecdn.com/invasionmetastasis-csbrp-161201085229/85/Invasion-metastasis-csbrp-24-320.jpg)






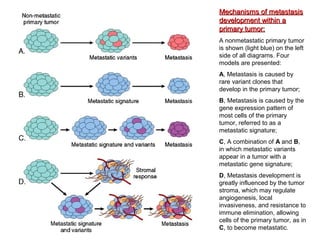



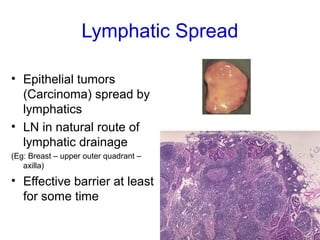
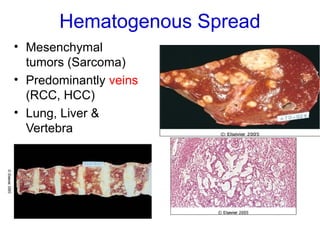







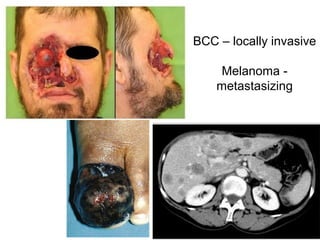
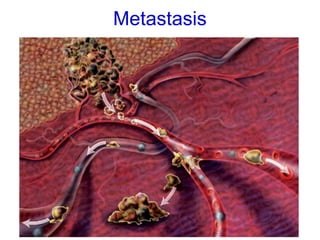
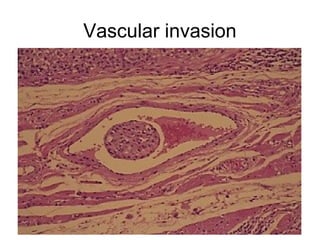
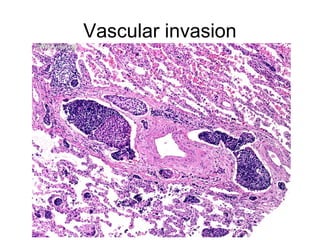
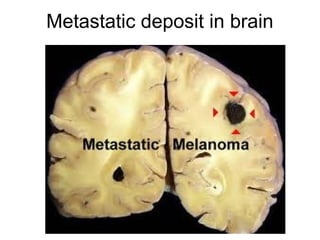
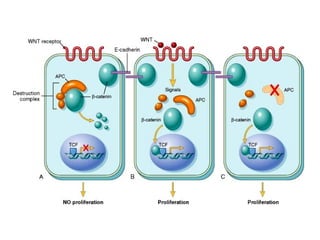
The document discusses the process of tumor invasion and metastasis. It describes how tumor cells must go through a series of steps called the metastatic cascade to break away from the primary tumor and form secondary tumors elsewhere. This involves invasion of the extracellular matrix and vascular dissemination. The key steps of ECM invasion are: 1) changes in cell adhesion, 2) degradation of the ECM, 3) attachment to novel ECM components, and 4) migration of tumor cells through active proteolysis and locomotion. Genetic changes can cause variations in metastatic potential between cancer types by promoting epithelial-to-mesenchymal transition or affecting signaling pathways. Tumors can spread via lymphatics, direct seeding of body cavities,























































































