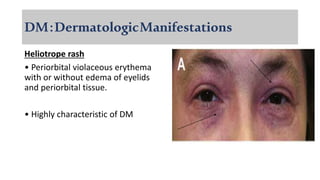
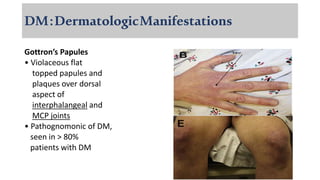
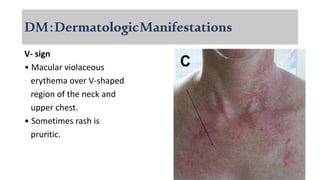
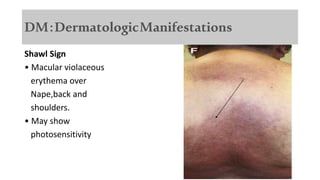
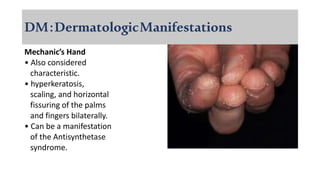
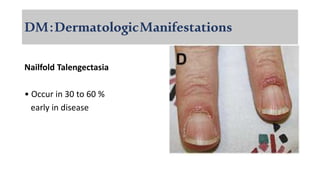
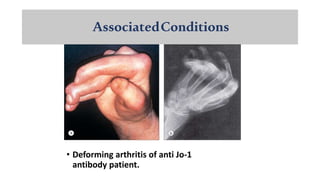
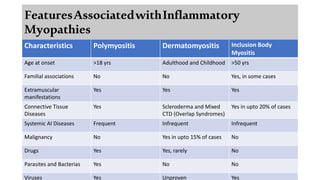
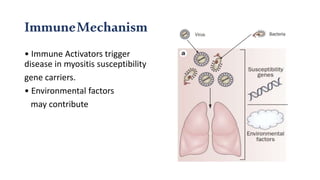
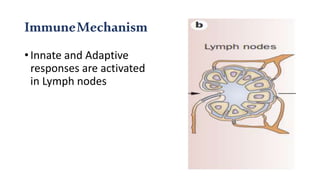
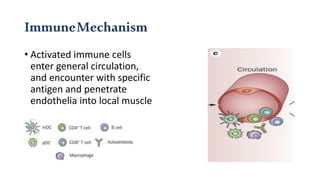
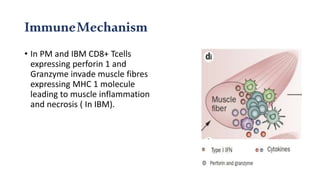
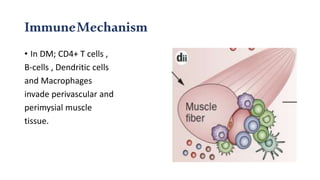
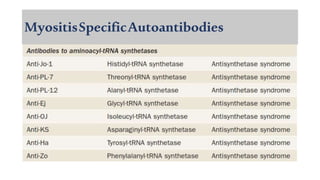
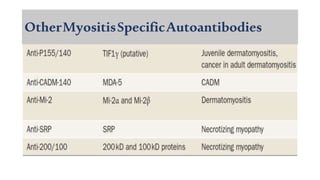
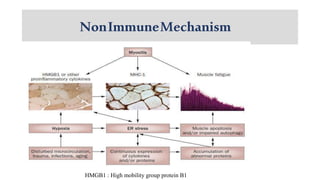
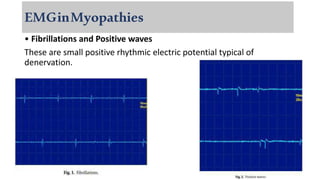
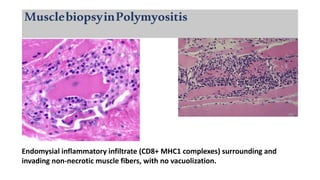
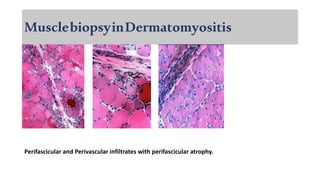
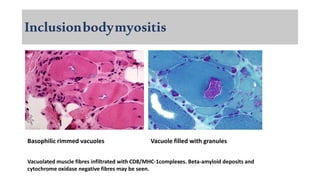
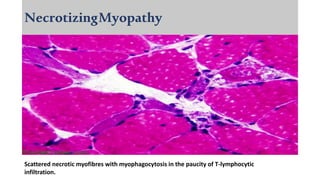
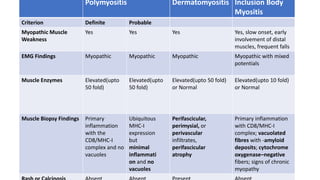
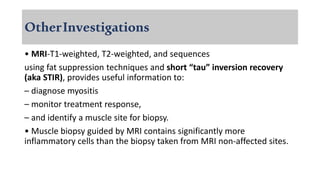
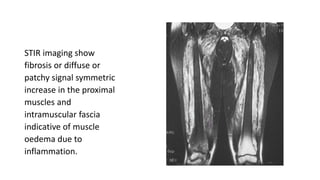
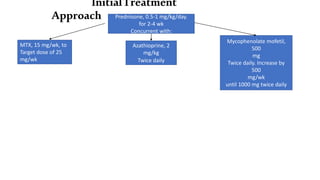
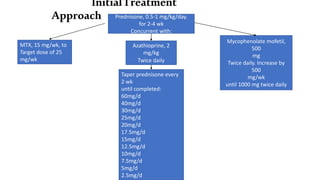
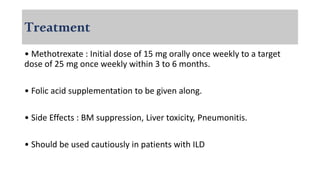
The document provides a comprehensive overview of inflammatory myositis, discussing its classification, clinical features, associated conditions, pathogenesis, diagnosis, and treatment strategies. It highlights the differences between types such as polymyositis, dermatomyositis, and inclusion body myositis, including their symptomatic presentations and demographic details. The document also outlines therapeutic approaches, emphasizing the importance of immunosuppressive therapies and monitoring for potential complications.