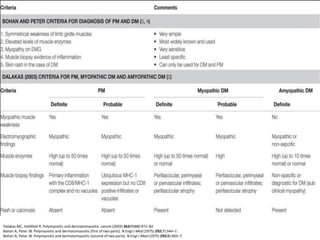
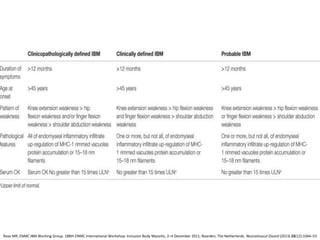
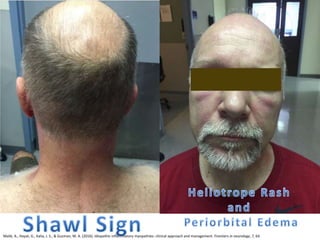
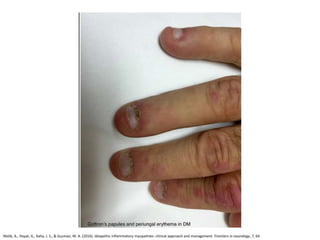
This document discusses idiopathic inflammatory myopathies (IIM), including their classification, clinical features, epidemiology, and relevant literature. The main types of IIM are polymyositis (PM), dermatomyositis (DM), necrotizing autoimmune myopathy (NAM), and sporadic inclusion body myositis (sIBM). DM is characterized by a skin rash in addition to muscle weakness. NAM presents with severe weakness but no rash. sIBM typically affects older males and causes both proximal and distal weakness. The document provides details on clinical manifestations and diagnostic criteria for each IIM subtype.




























































































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)


















