Downloaded 440 times























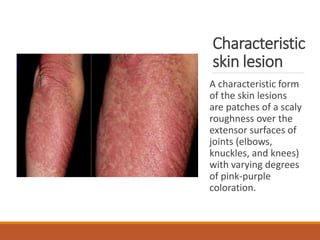
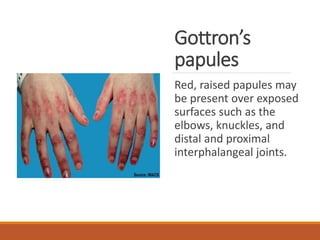
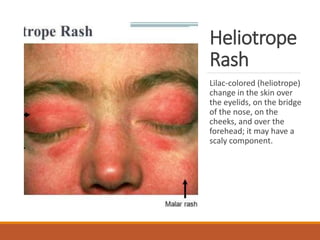
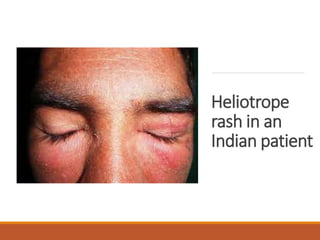


















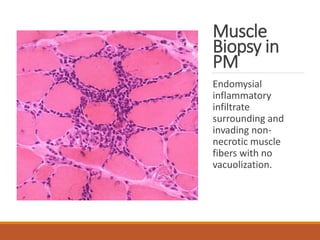
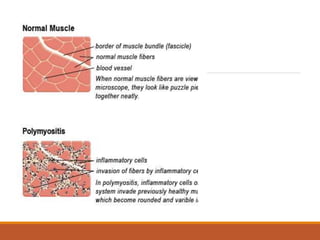
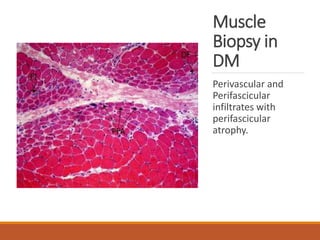
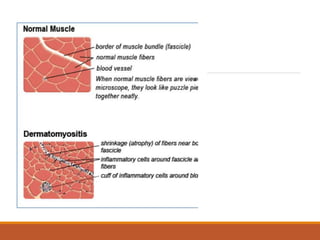
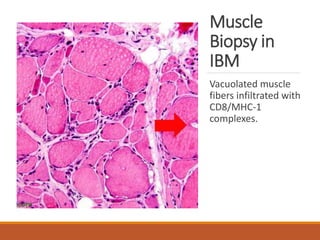
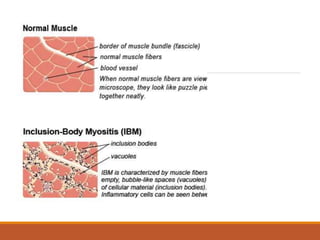





















This document provides an overview of idiopathic inflammatory myositis, which includes three main types: polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM). PM primarily involves muscle weakness while DM also includes skin involvement. IBM predominantly affects those over 50. Together these conditions represent acquired causes of skeletal muscle weakness. The document discusses their definitions, classifications, epidemiology, clinical features, pathogenesis, associations and treatment considerations.











































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)