Downloaded 94 times



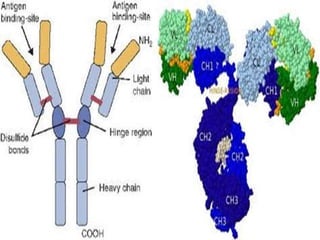




![D) Hyper-IgM syndromes (including deficiencies of
CD40 ligand (CD154), activation-induced cytidine
deaminase [AID], and uracil-nucleoside-glycosylase
[UNG]):
This is a heterogeneous group of disorders in
which normal or elevated IgM levels are found along
with low levels of IgA, IgG, and, sometimes, IgE.
E) Common variable immunodeficiency (CVID)
CVID is usually differentiated from XLA and
autosomal recessive agammaglobulinemia by the
presence of B-cells, visible tonsils or a history of
tonsillectomy, and palpable or even enlarged lymph
nodes.](https://image.slidesharecdn.com/hypogammaglobulinemia-150526194011-lva1-app6892/85/Hypogammaglobulinemia-10-320.jpg)





Hypogammaglobulinemia is characterized by a decrease in plasma gamma globulins due to a deficiency in B lymphocytes. It can be caused by primary (congenital) disorders including X-linked agammaglobulinemia, autosomal recessive agammaglobulinemia, specific antibody deficiency, and hyper-IgM syndromes. Secondary causes include nephrotic syndrome, protein-losing enteropathy, and immunosuppressive therapy. Treatment involves immunoglobulin replacement therapy through vaccination or bone marrow transplantation for certain syndromes.





















































































