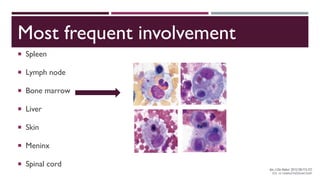
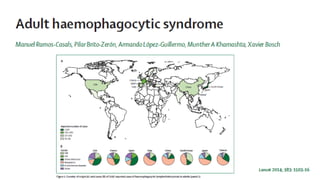
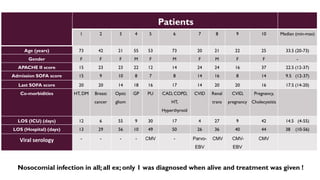
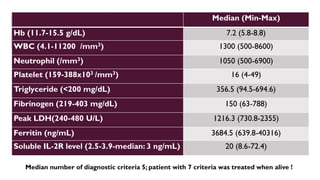
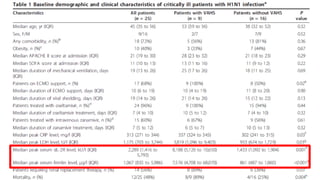
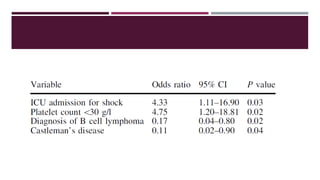
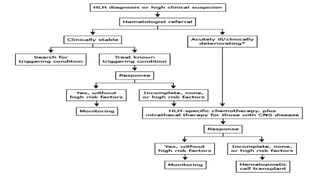
Hemophagocytic lymphohistiocytosis (HLH) is a rare and life-threatening syndrome characterized by uncontrolled activation of macrophages and lymphocytes, leading to excessive inflammation and organ damage. It can occur primarily due to genetic mutations or secondarily in association with infections, malignancies, or rheumatologic conditions. The pathophysiology involves defective cytotoxic lymphocyte function and cytokine overproduction. HLH is diagnosed based on clinical and laboratory criteria including prolonged fever, cytopenias, hypertriglyceridemia/hypofibrinogenemia, hyperferritinemia, and hemophagocytosis. Early diagnosis and treatment with chemotherapy, steroids, and hematopoietic stem cell transplantation is needed for survival, as H