Downloaded 393 times





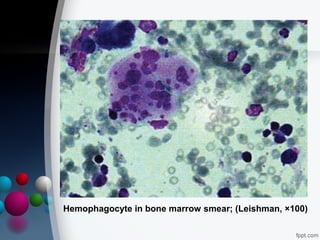
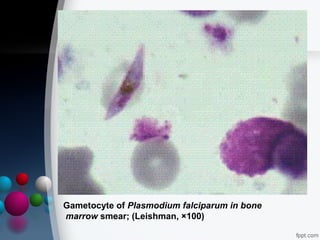



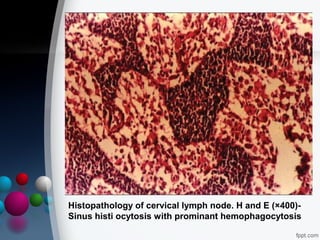
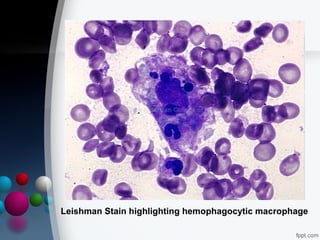
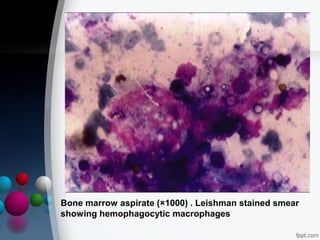

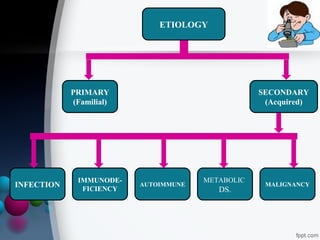







This document presents two case reports of hemophagocytic syndrome (HPS). HPS is a rare disorder characterized by uncontrolled phagocytosis of blood cells in the bone marrow. It can be secondary to infections, malignancies, autoimmune diseases, or immunodeficiencies. The first case report describes an 11-month old child with HPS associated with Plasmodium falciparum malaria infection. The second case report involves a 17-year old male with HPS complicating tuberculosis infection. Both patients exhibited cytopenias, hepatosplenomegaly, and hemophagocytosis identified in bone marrow samples. The malaria patient recovered with antimalarial treatment, while the tuberculosis patient responded to antitub


























































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)








