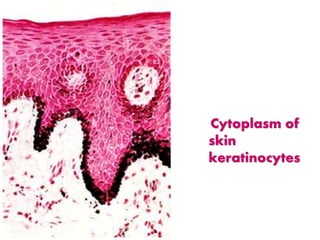
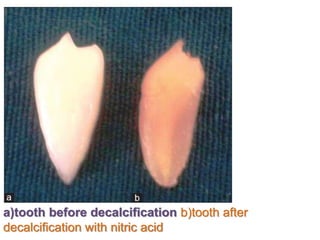
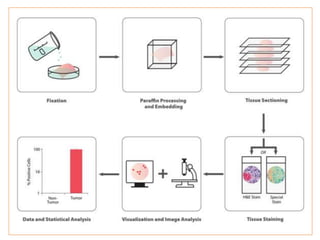
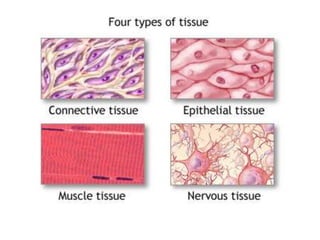
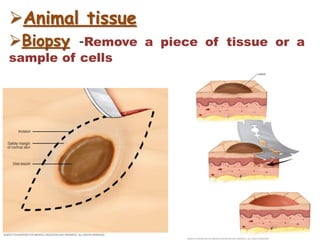
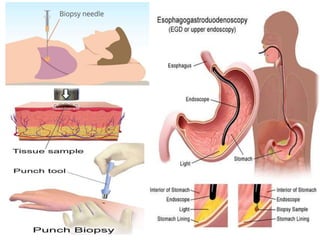
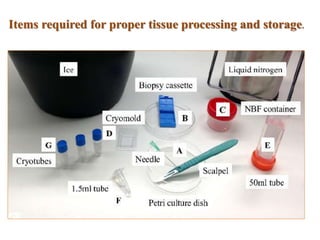
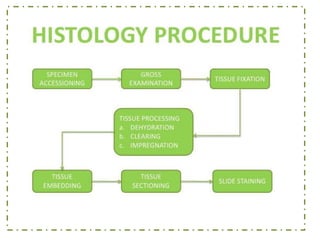
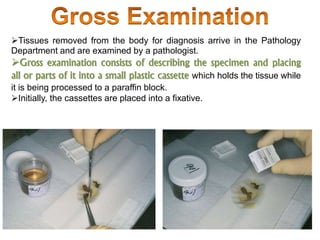
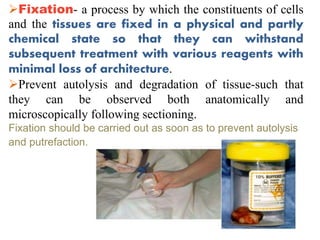
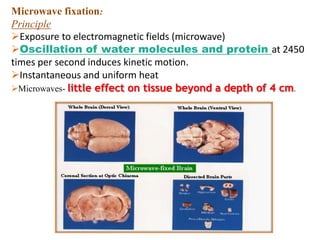
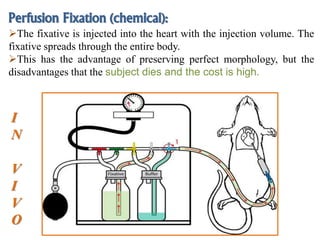
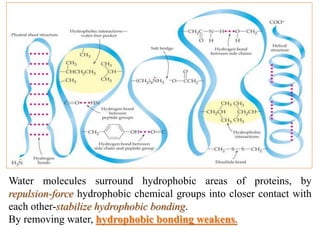
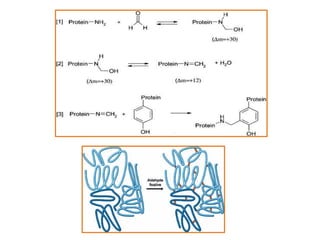
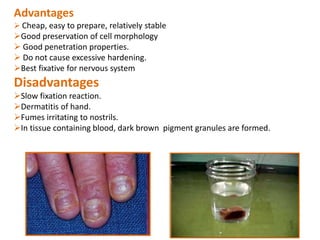
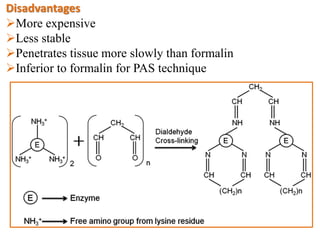
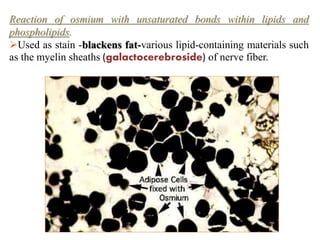
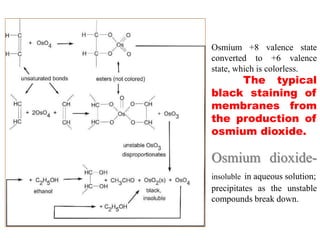
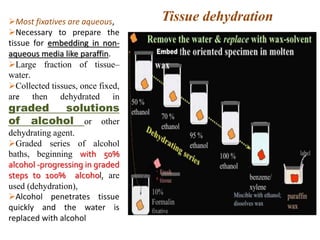
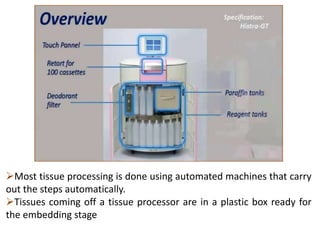
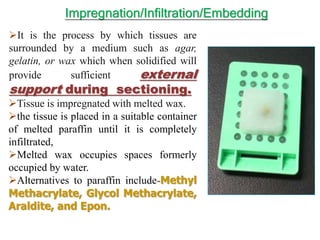
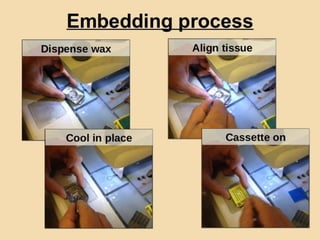
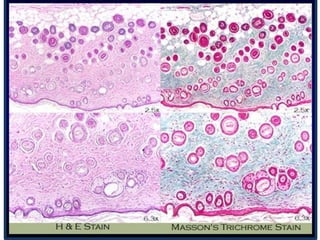
Histopathology examines minute tissue alterations from disease. Samples come from cadavers, autopsies, animal tissues, or biopsies. Histopathological examination is useful for establishing disease pathogenesis and diagnosing diseases that are difficult to diagnose by other means. It typically begins with surgery or biopsy to collect tissue samples, which are then fixed, processed, and examined microscopically. Common fixatives include formaldehyde and glutaraldehyde, which cross-link proteins to preserve tissue morphology and prevent autolysis.

















































![Steps
Oxidation (converts sugars -OH to –CHO), by phosphomolybdic acid or
KMnO₄.
Sensitization- treated with Ferric ammonium sulfate.
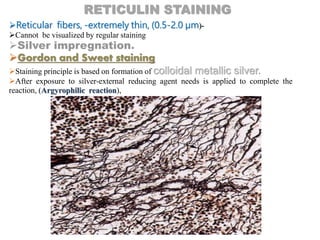
(Reticulin fibers- little affinity for silver)
Impregnation -with AgNO₃ or [Ag(NH₃)₂]+,
Sensitized sites accept silver.
Reduction- tissue sections-uncolored-reducing agent HCHO
HCHO + Ag -complex HCOOH + Ag (s) (brown-
black)
Contrasting
Ag deposits (brown-black) ----- AuCl₃ (purple-black)
Removal of by adding sodium thiosulfate.
Counterstain- nuclear fast red.
Study architecture of parenchymal organs
Study benign and malignant bone marrow disorders](https://image.slidesharecdn.com/histopathologicaltechnique-160507124837-180504104450/85/Histopathological-techniques-sectioning-STAINING-EMBEDDING-fixaton-microtomy-85-320.jpg)