Downloaded 26 times


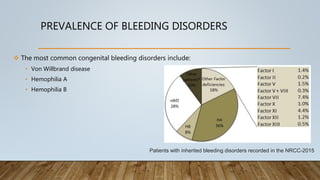








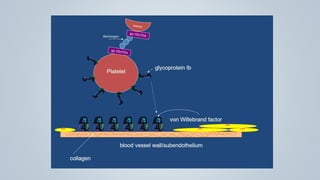










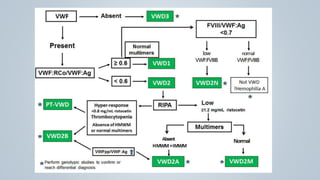



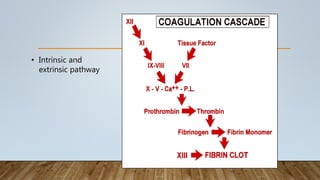
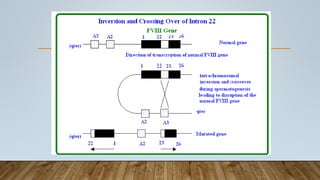


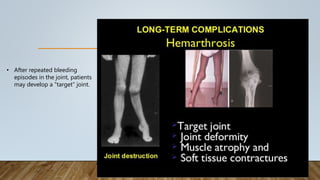
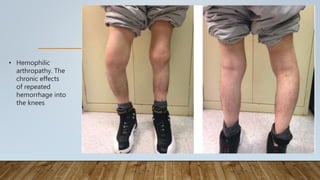


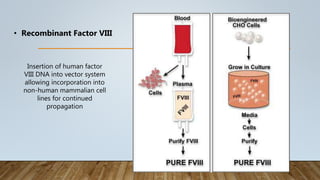








This document provides an overview of inherited bleeding disorders, focusing on von Willebrand disease, hemophilia A, and hemophilia B. It classifies inherited bleeding disorders and discusses their prevalence. For von Willebrand disease, it describes the classification, etiology, functions of von Willebrand factor, diagnosis of different types, and management. It also discusses the formation of the primary hemostatic plug. For hemophilia A and B, it covers clinical manifestations, diagnosis, and management, including use of factor replacement therapies.























































































