Download to read offline








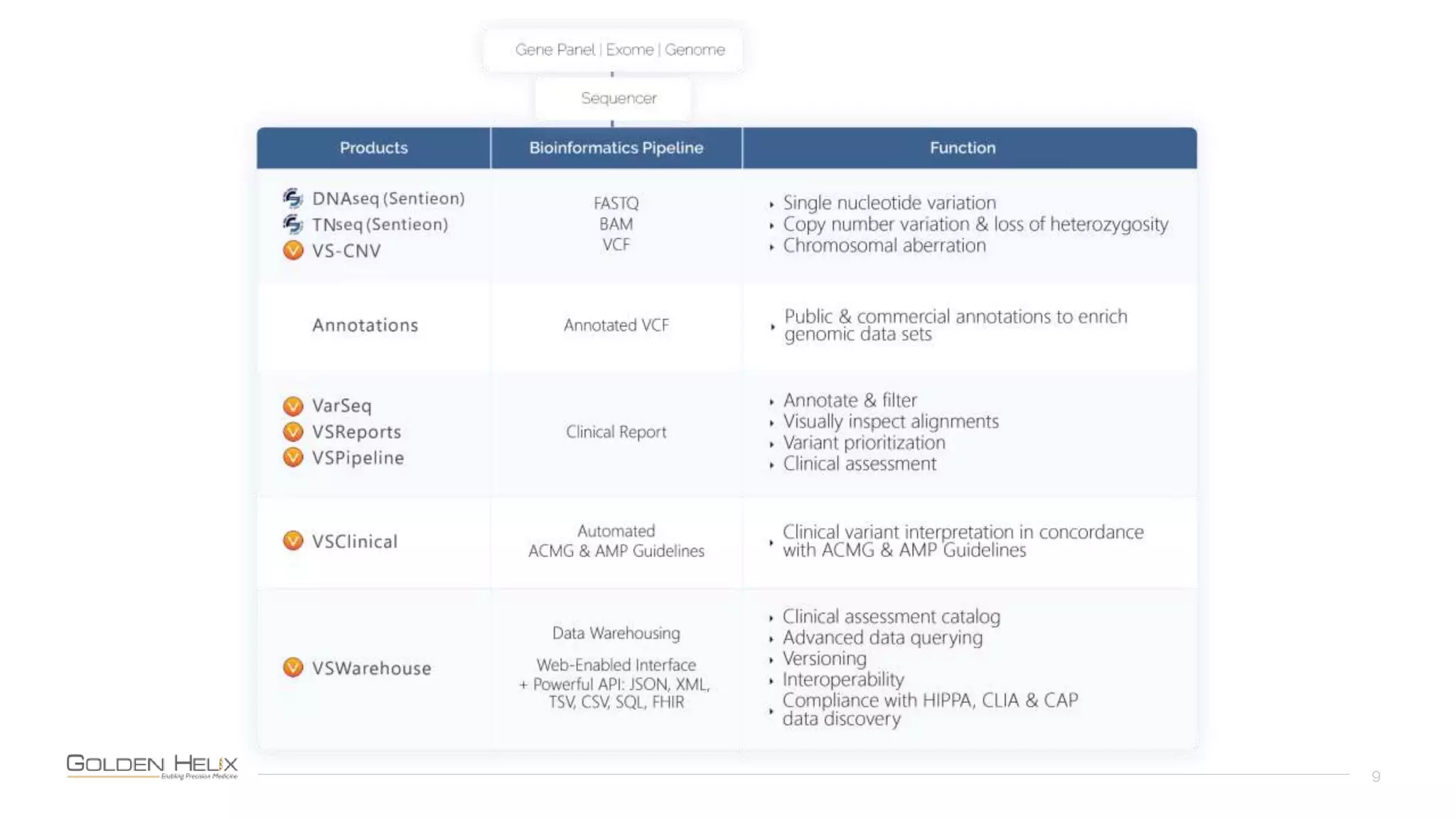
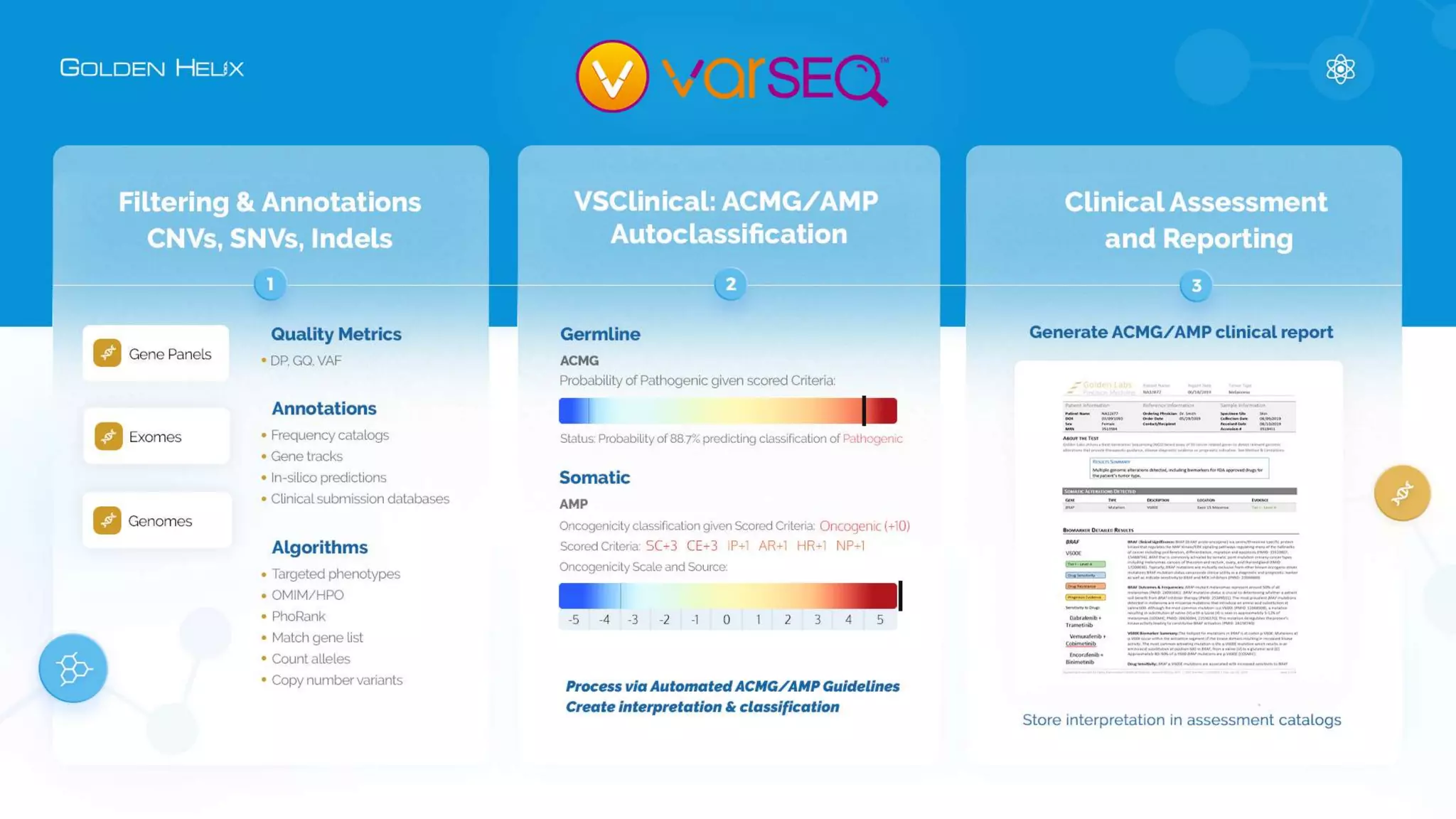


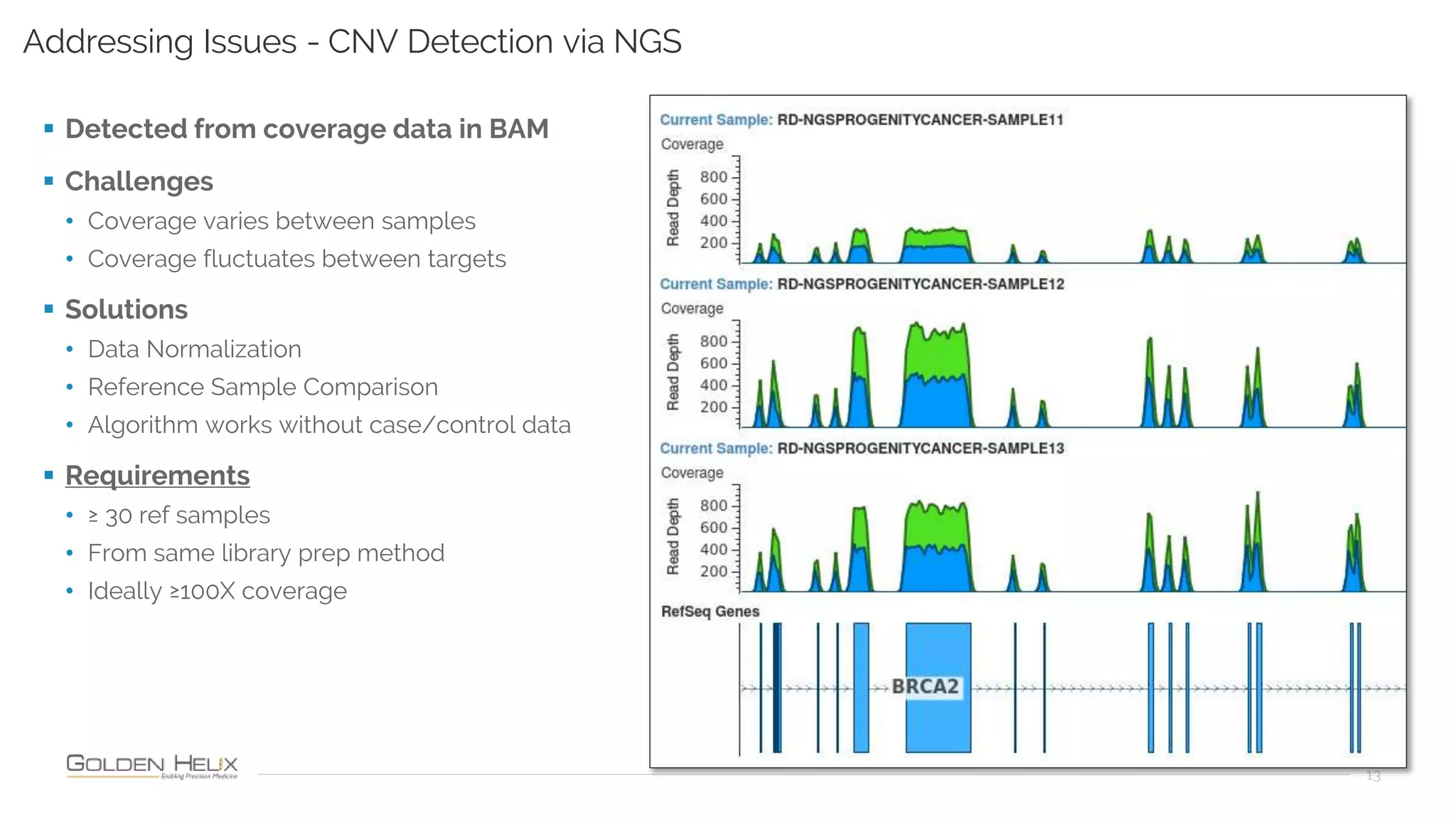
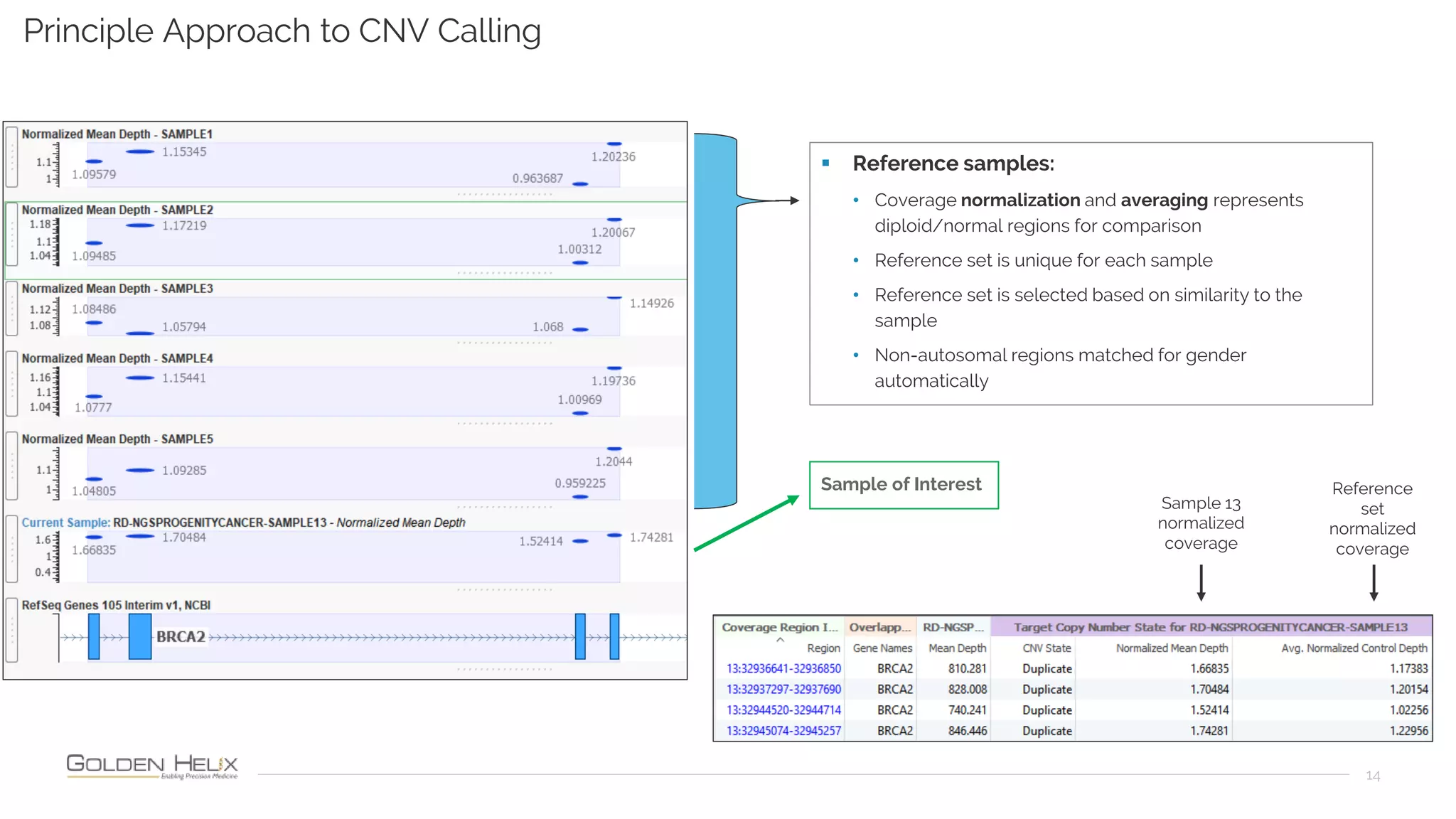
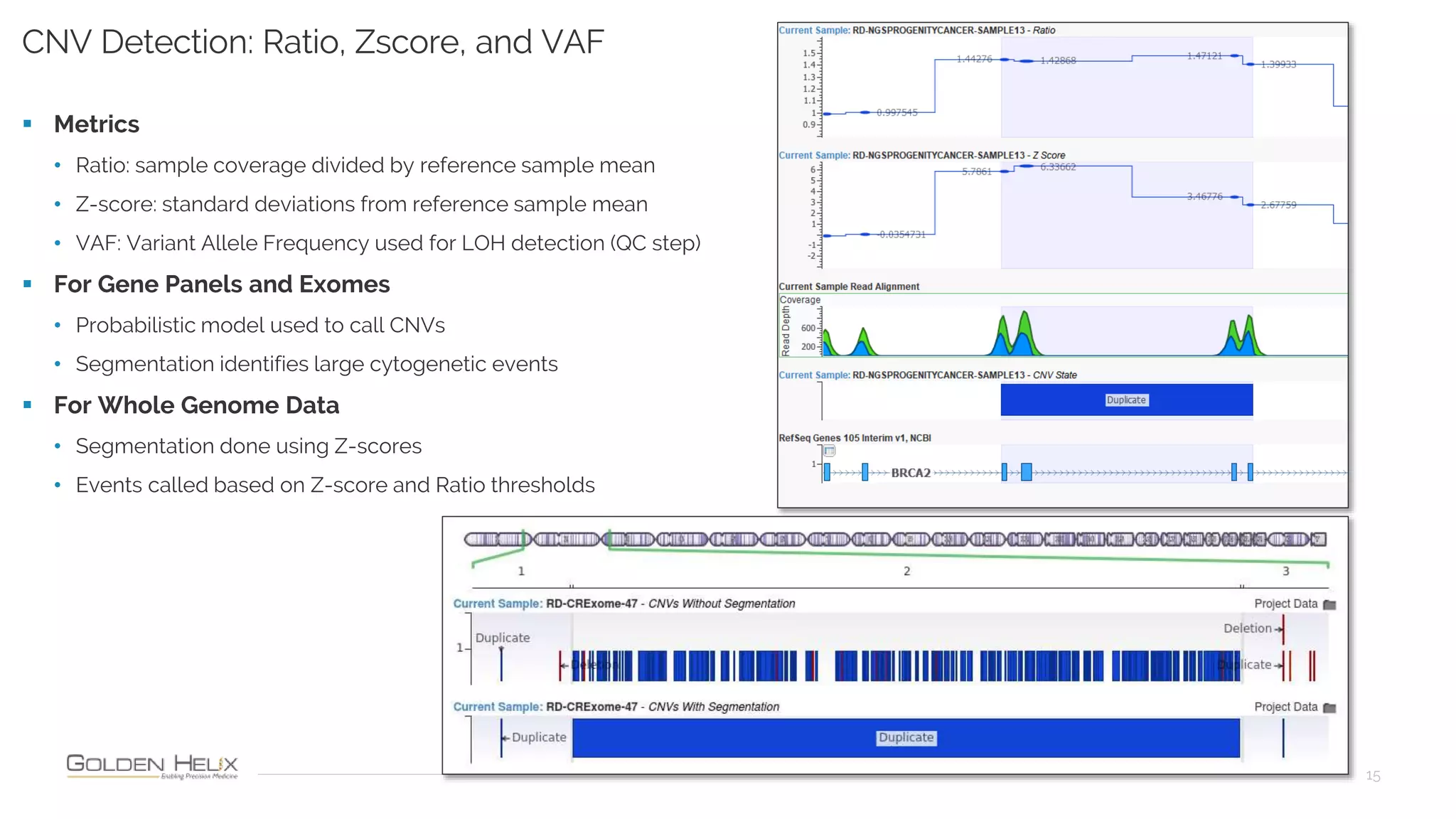
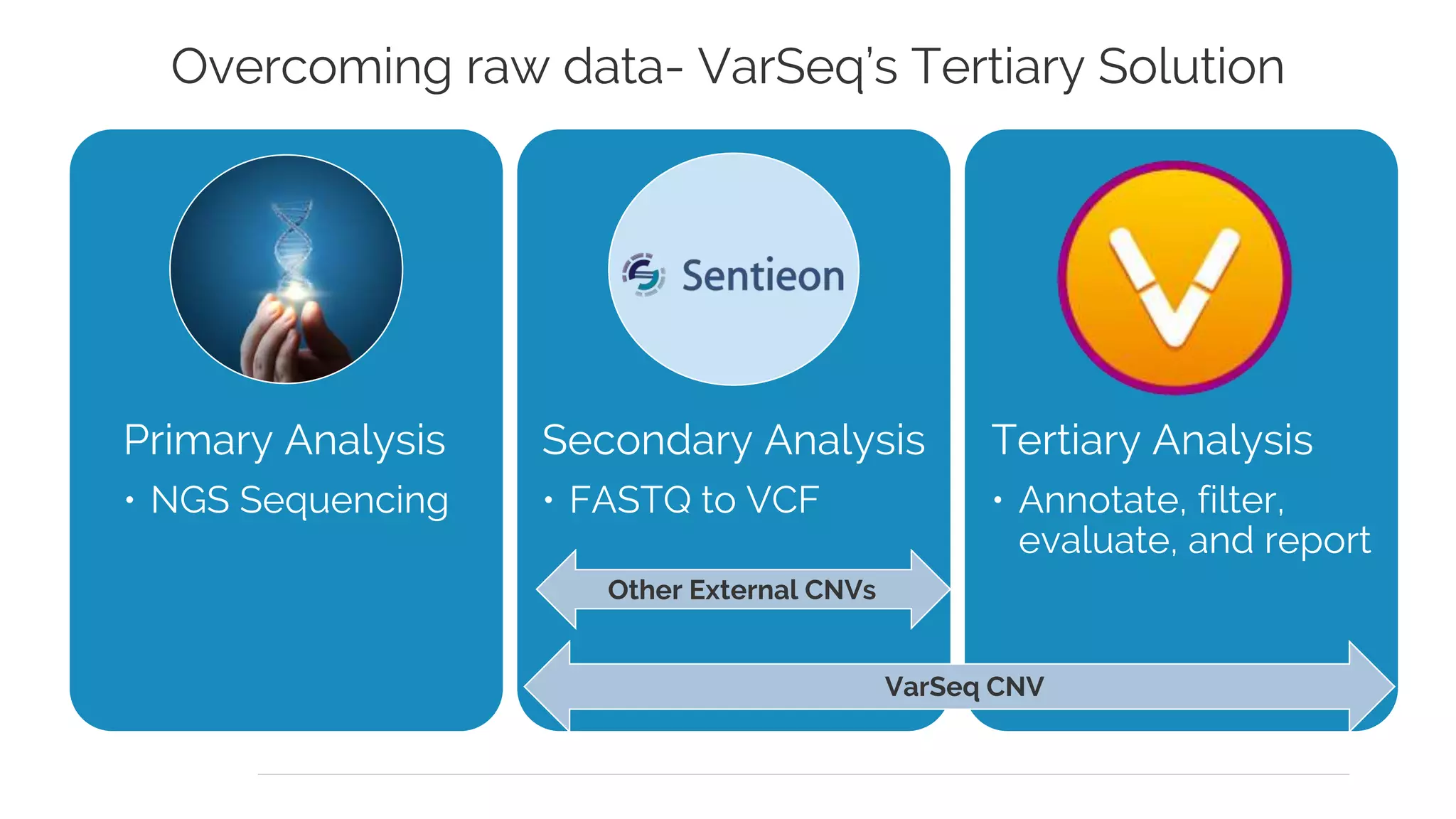
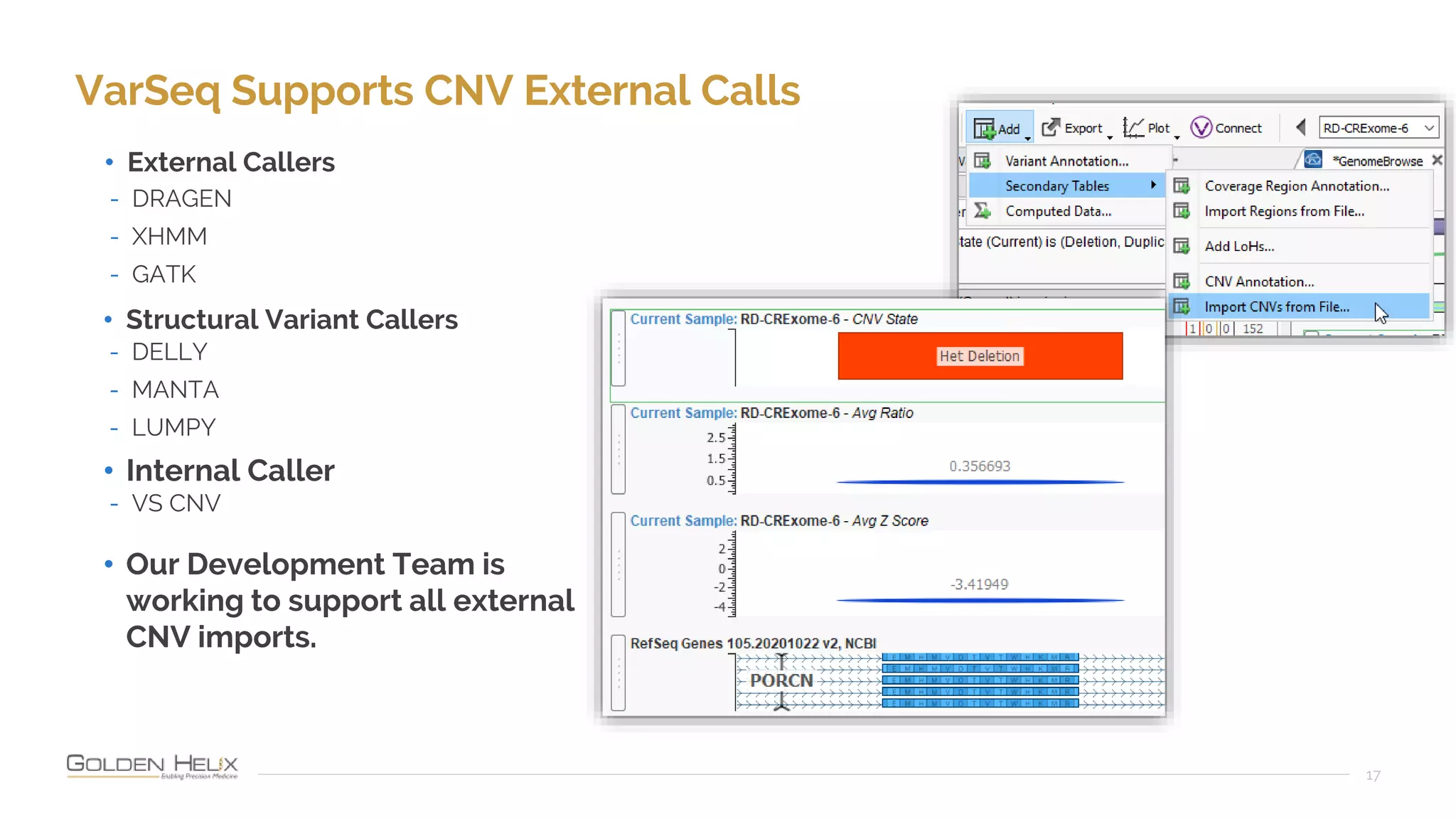
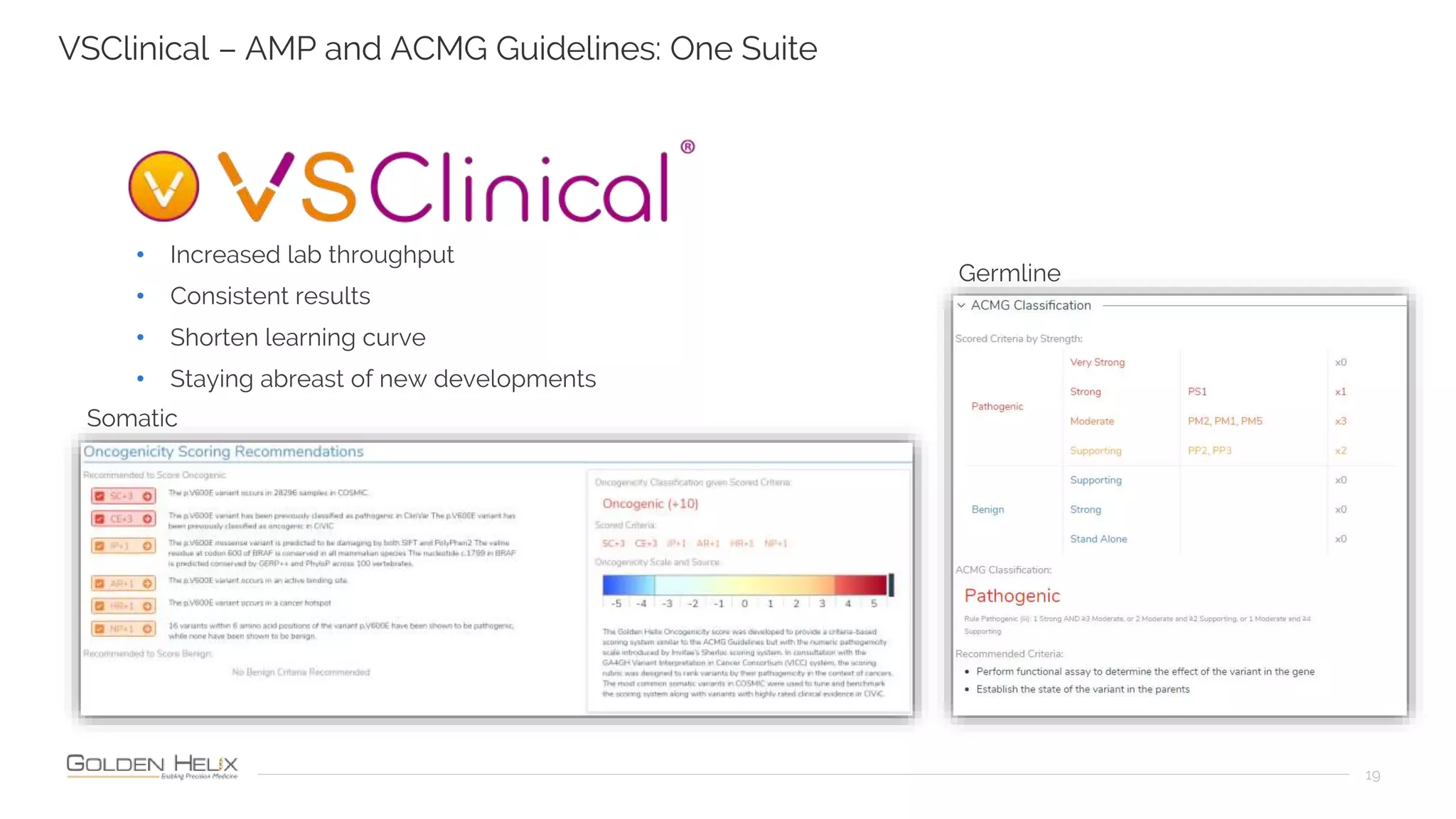
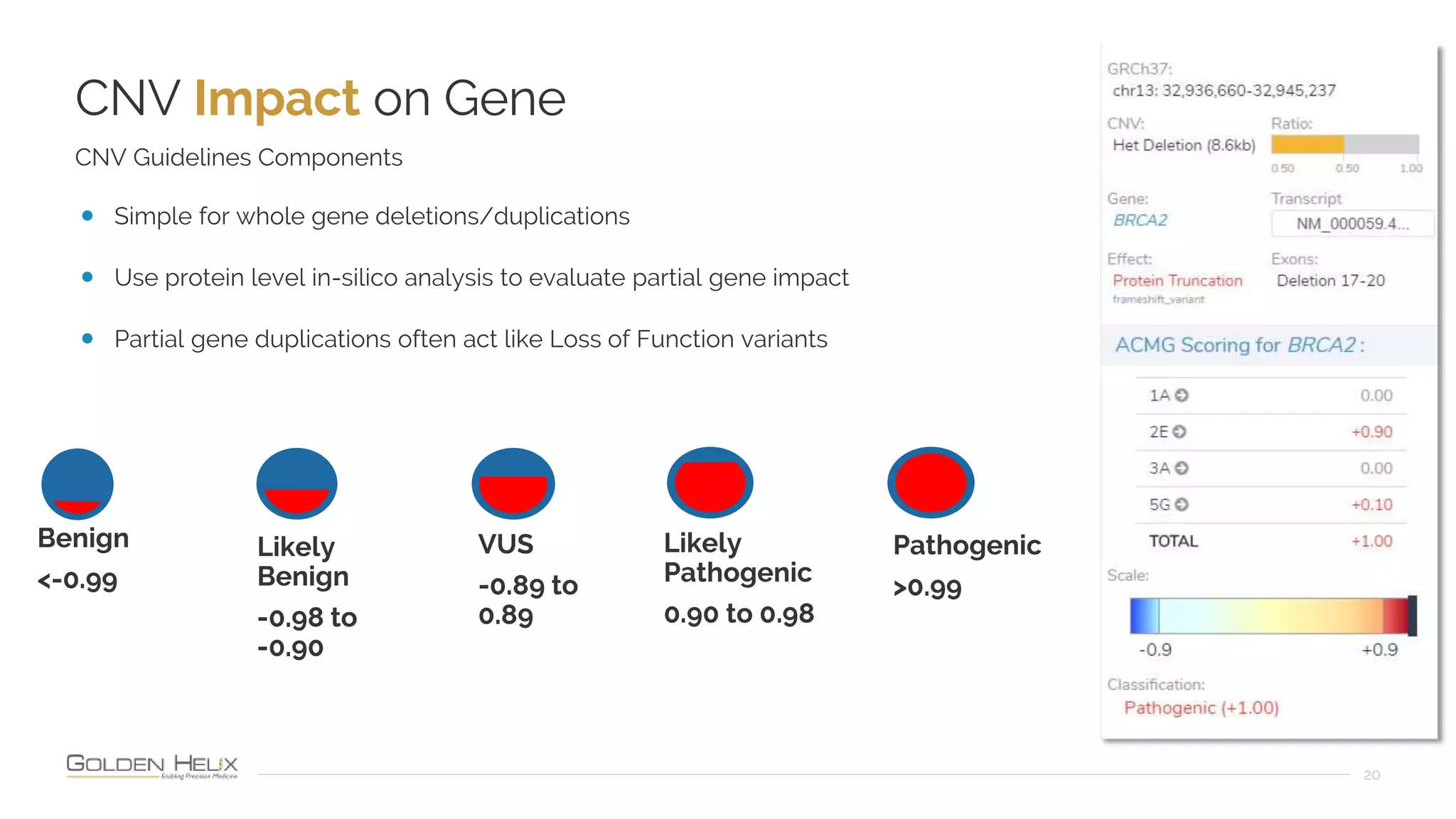







Golden Helix is presenting on their software VarSeq, which can handle a variety of CNV caller inputs. VarSeq uses normalization and reference sample comparison to call CNVs from NGS coverage data. It can detect small, medium, and large CNVs from gene panels, whole exomes, and whole genomes. Previous customers have validated VarSeq for these applications. The presentation demonstrates VarSeq's CNV detection approach and ability to import calls from external callers. It also discusses using VarSeq with their guidelines software to evaluate CNV impact according to ACMG/AMP standards.























































































