Download to read offline






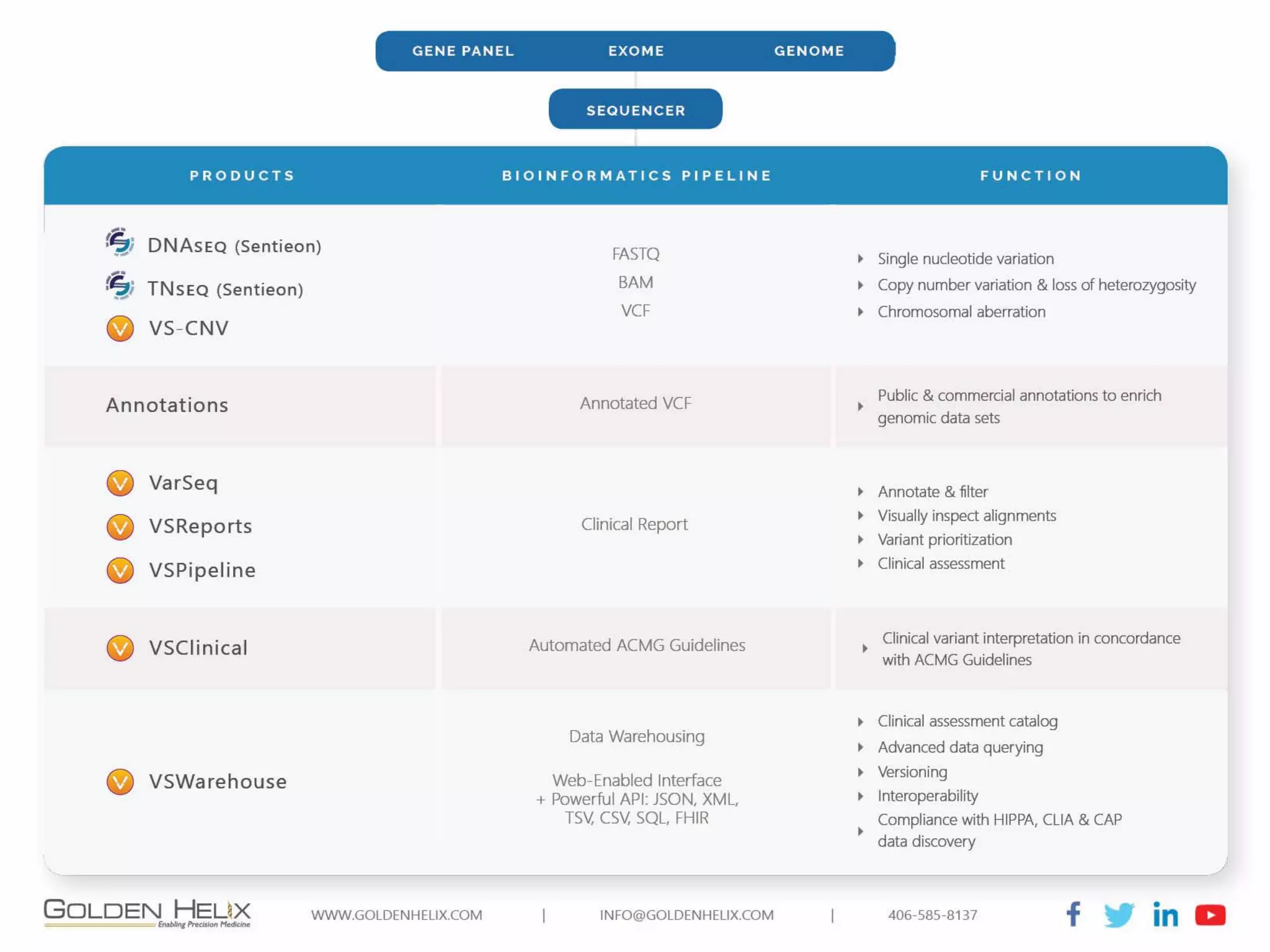

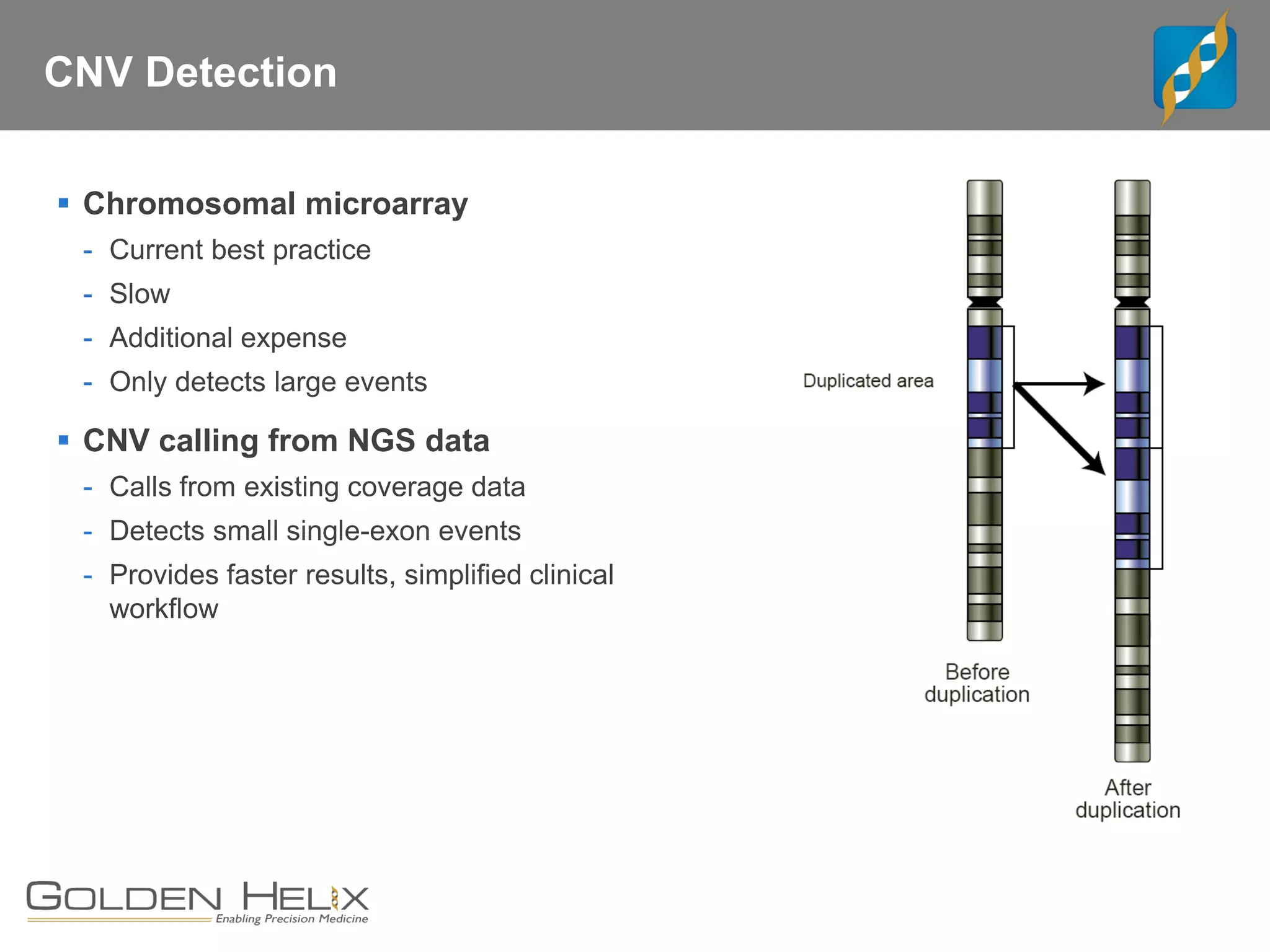
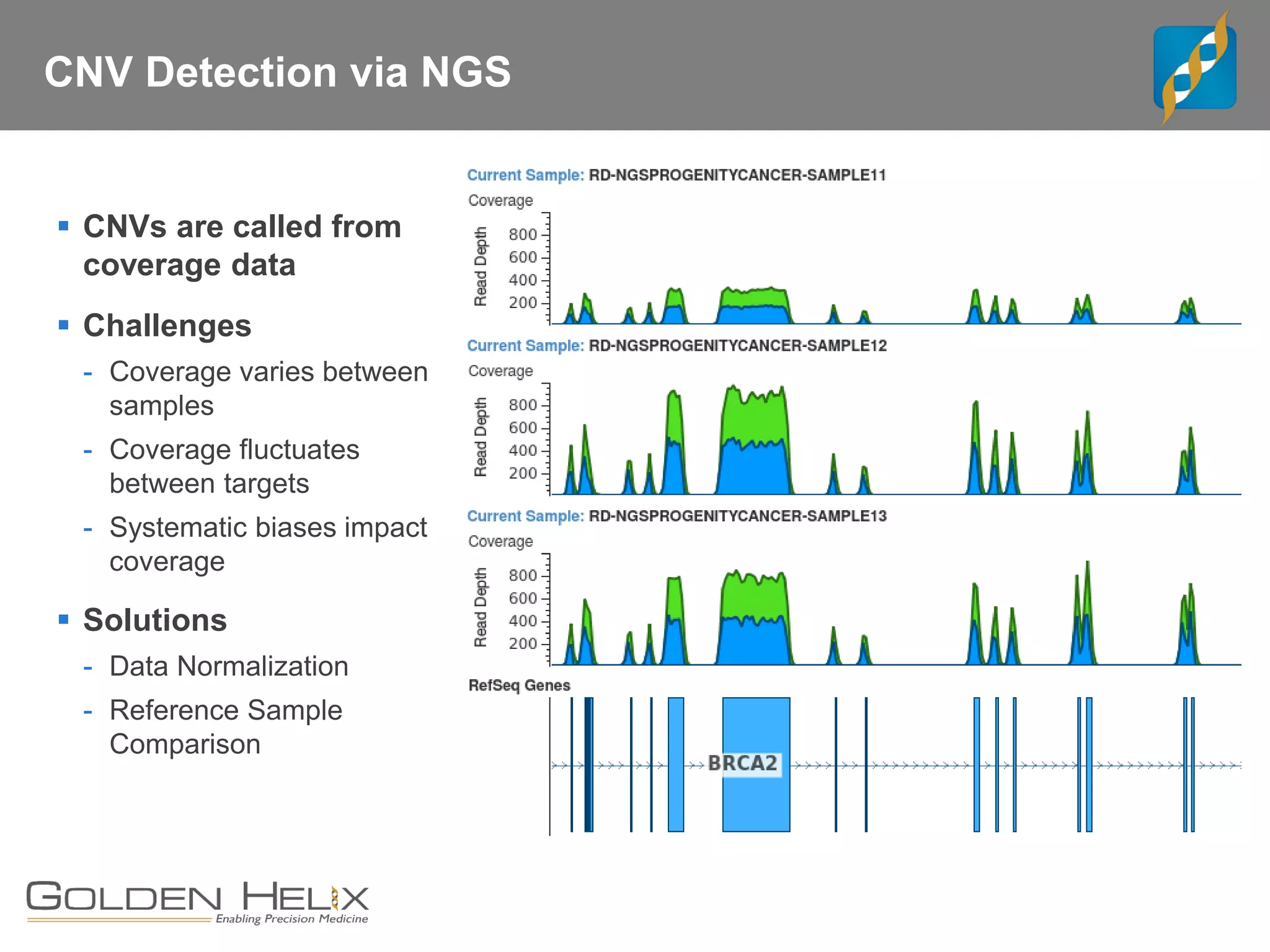
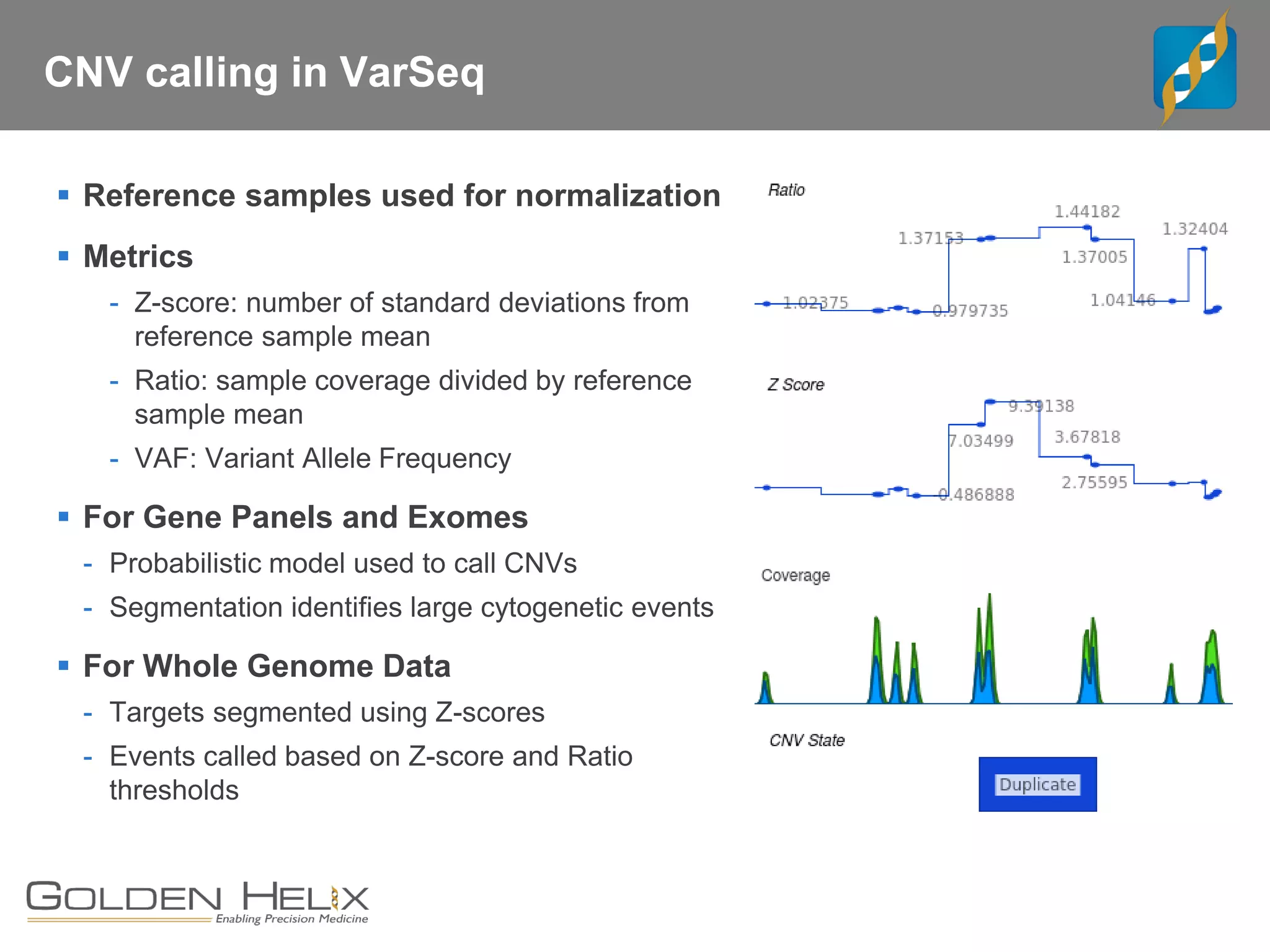



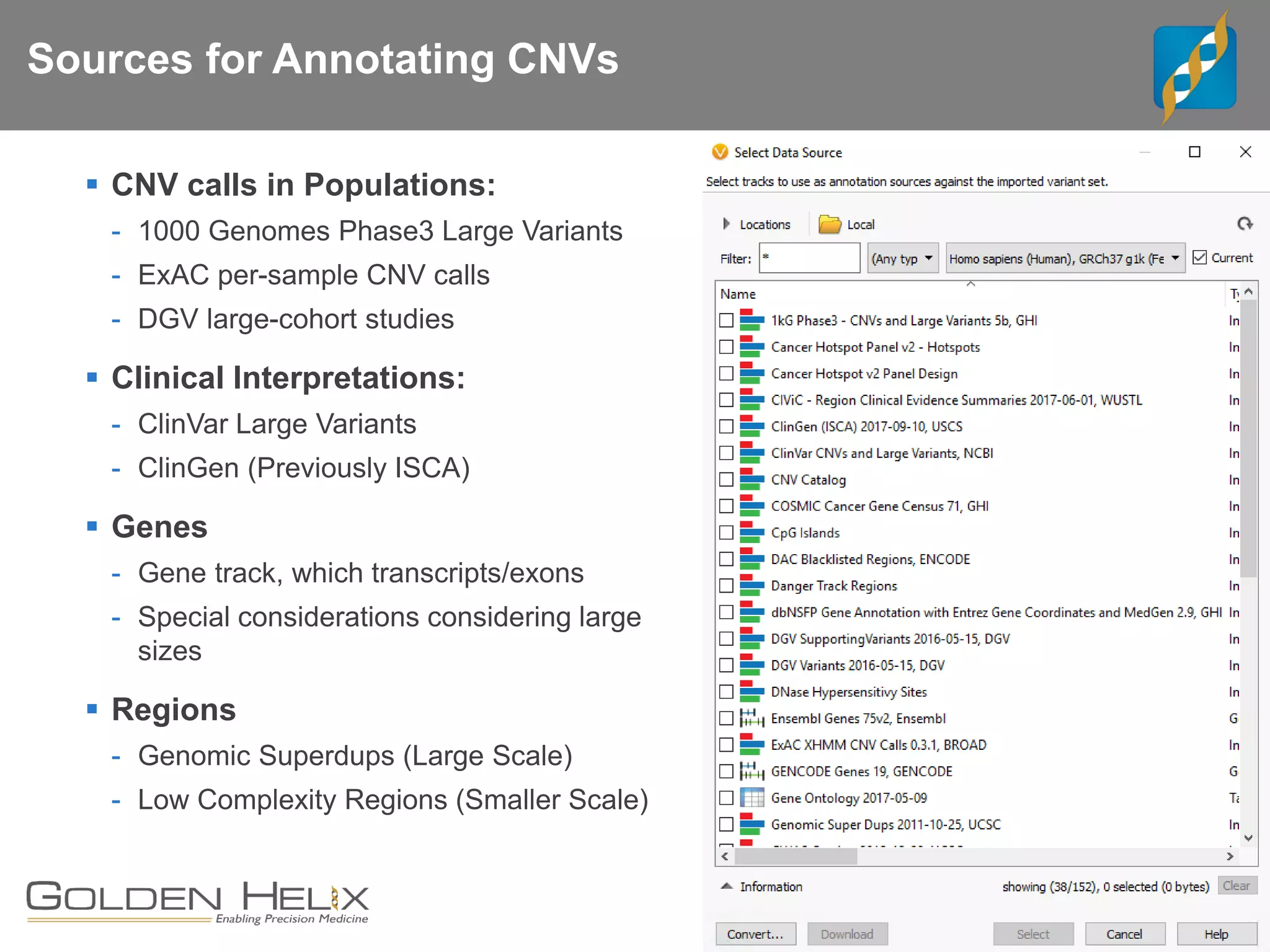







Golden Helix offers software solutions for analyzing copy number variations (CNVs) detected from next-generation sequencing (NGS) data. Their VarSeq software uses normalization, reference samples, and probabilistic modeling to call CNVs from gene panels, exomes, and whole genomes. It flags low quality CNV calls and samples based on coverage and variability. VarSeq annotates CNVs by comparing to public databases and applying the Jaccard index metric to determine similarity between regions. Golden Helix will demonstrate VarSeq at the AMP conference in November 2018.












































































