Downloaded 12 times








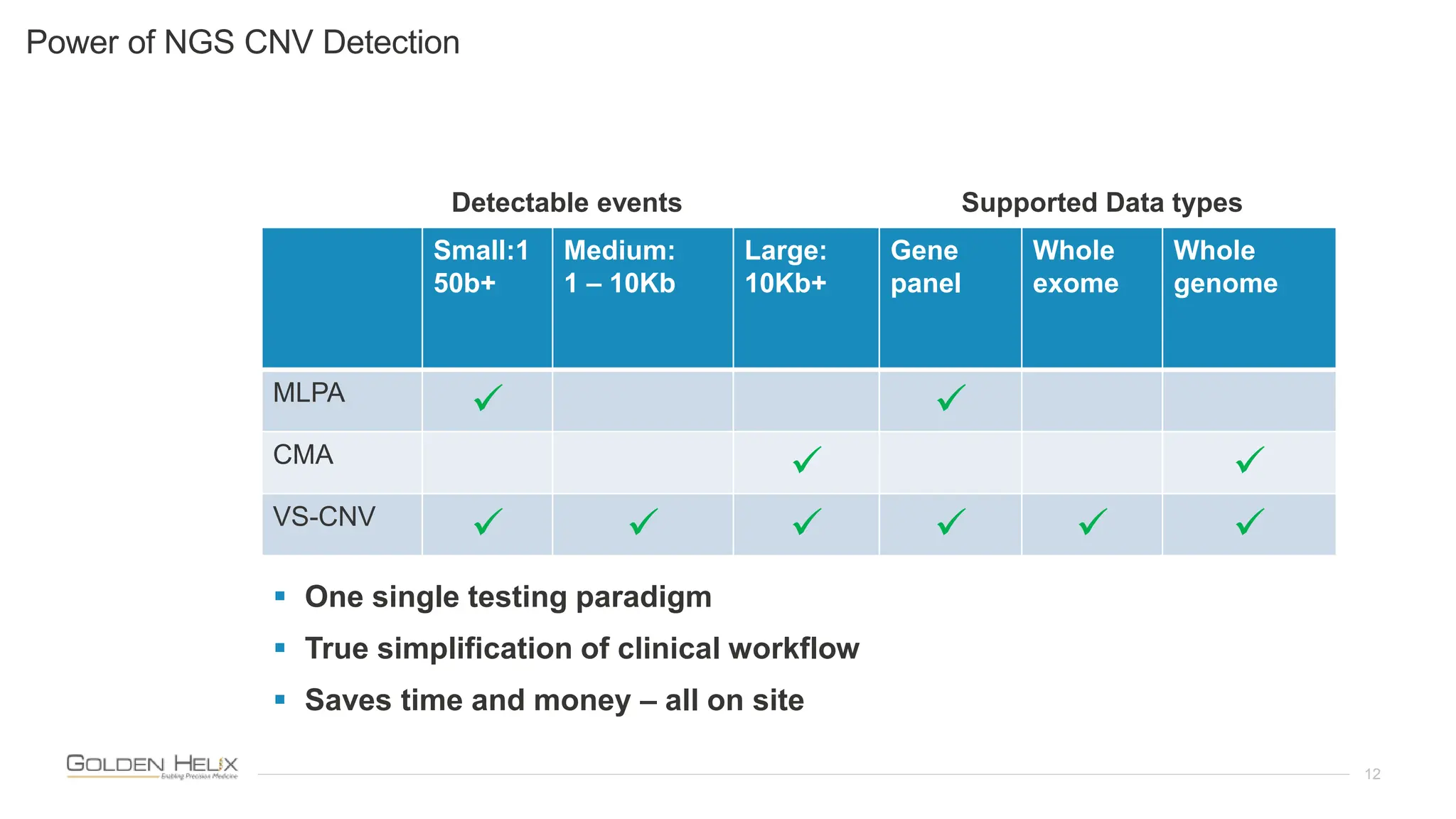
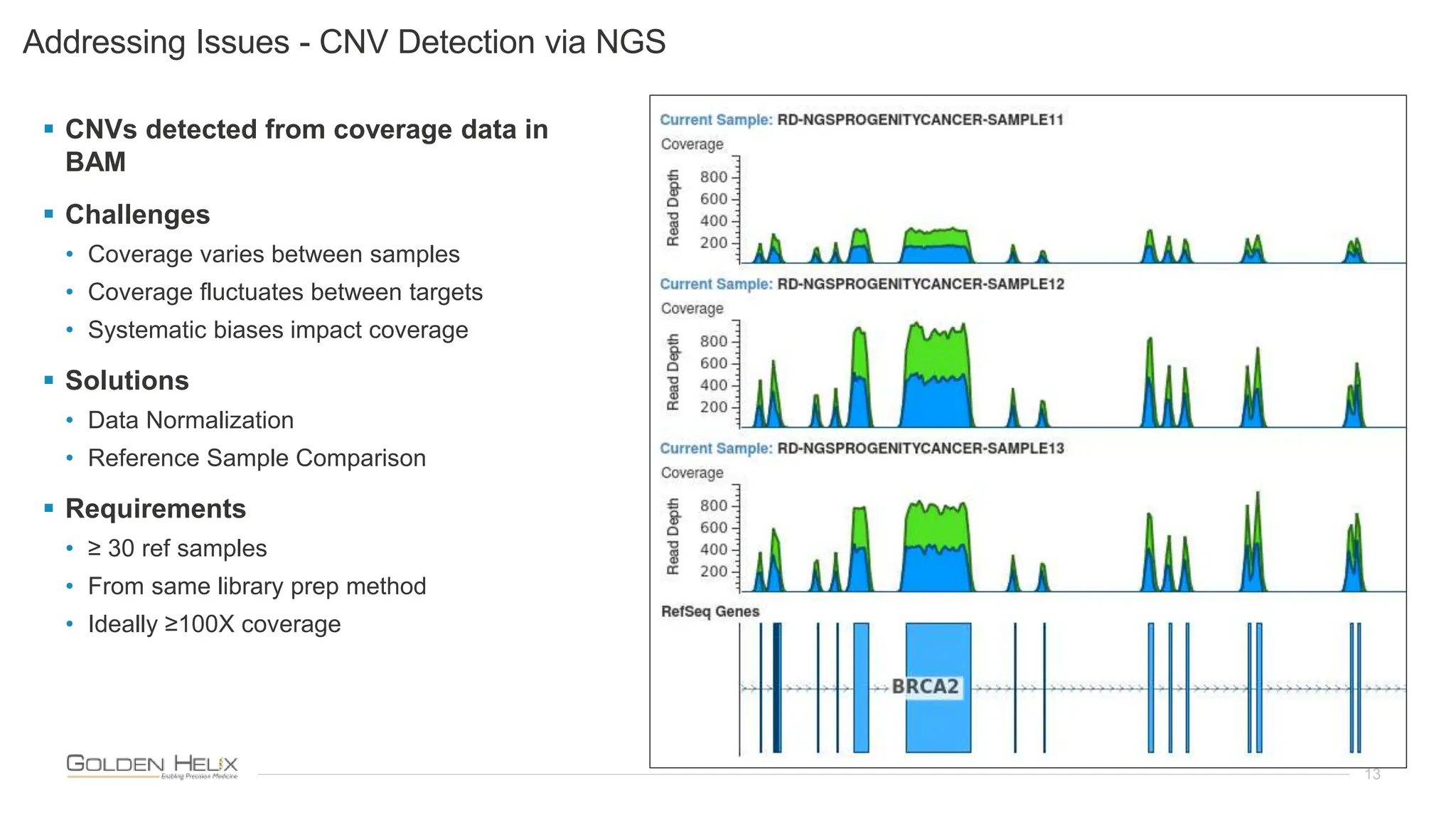
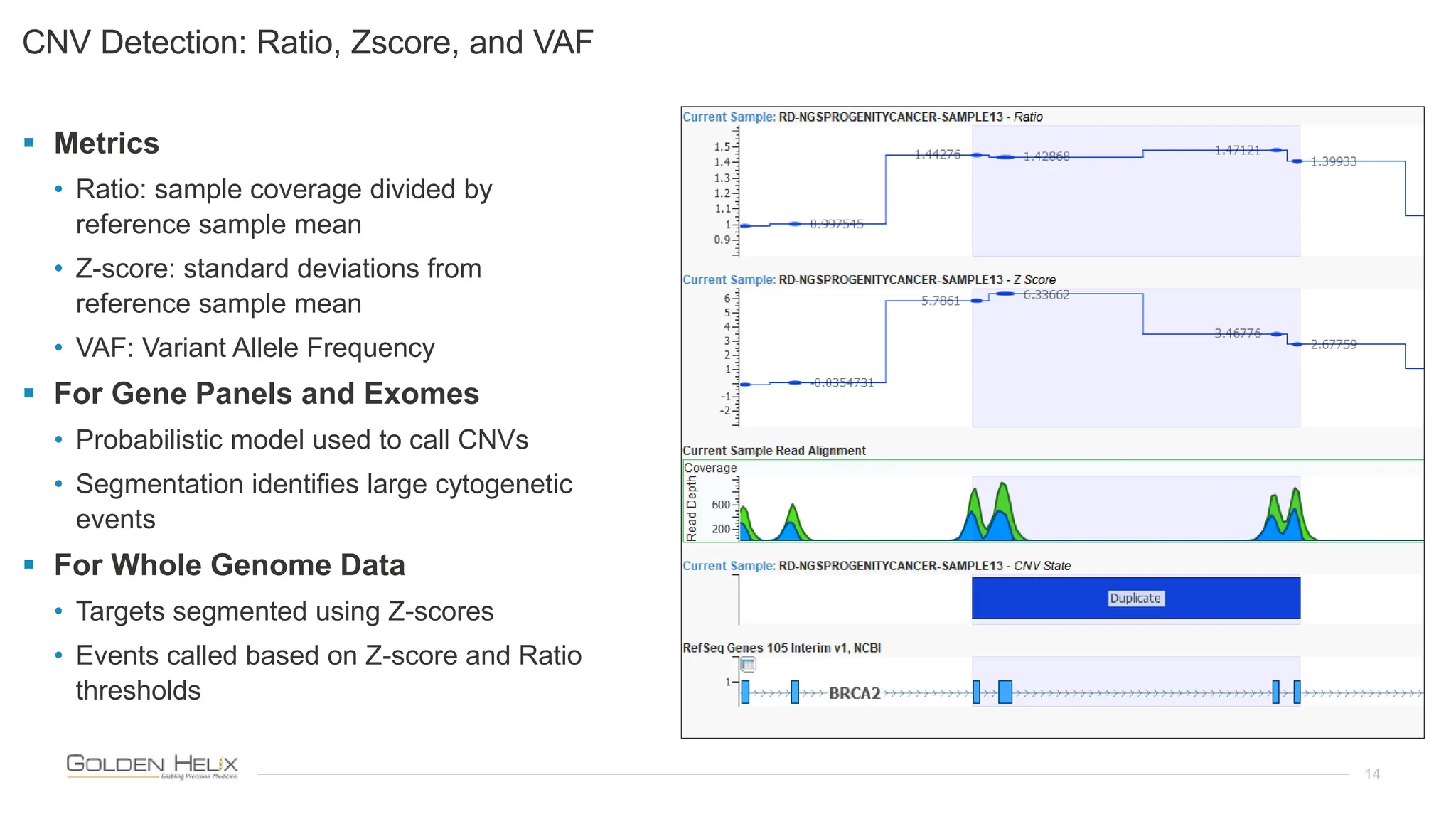






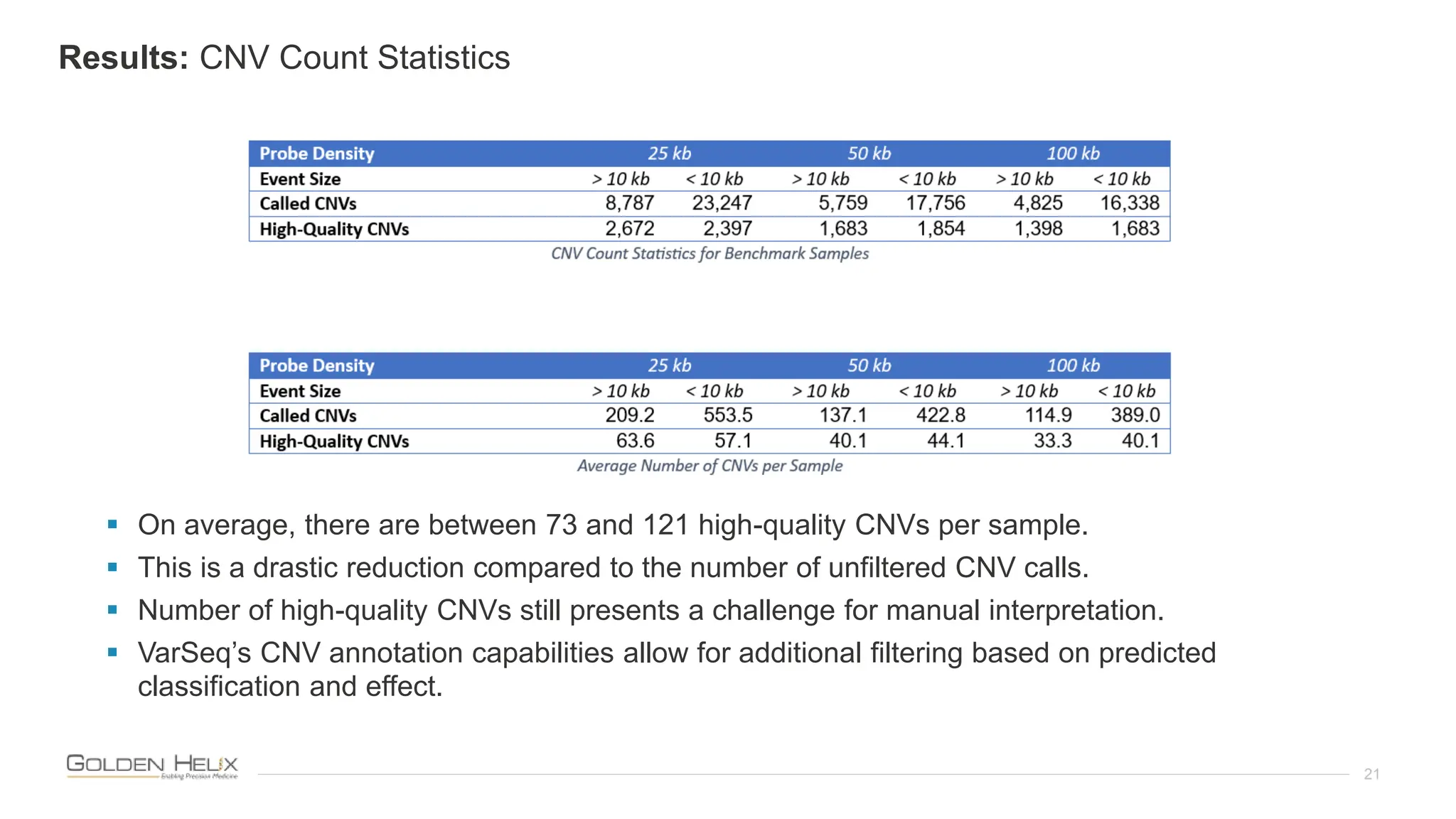







The document presents an analysis of the Twist Exome with CNV backbone using varying probe densities, highlighting its effectiveness in CNV detection through the VS-CNV algorithm. Funded by multiple NIH grants, the research involved evaluating sensitivity across three probe densities with a benchmark dataset, revealing a 100% sensitivity for the algorithm. Additionally, the document discusses the importance of quality management in bioinformatics with ISO certification and invites participation in upcoming user meetings and innovation awards.



































































