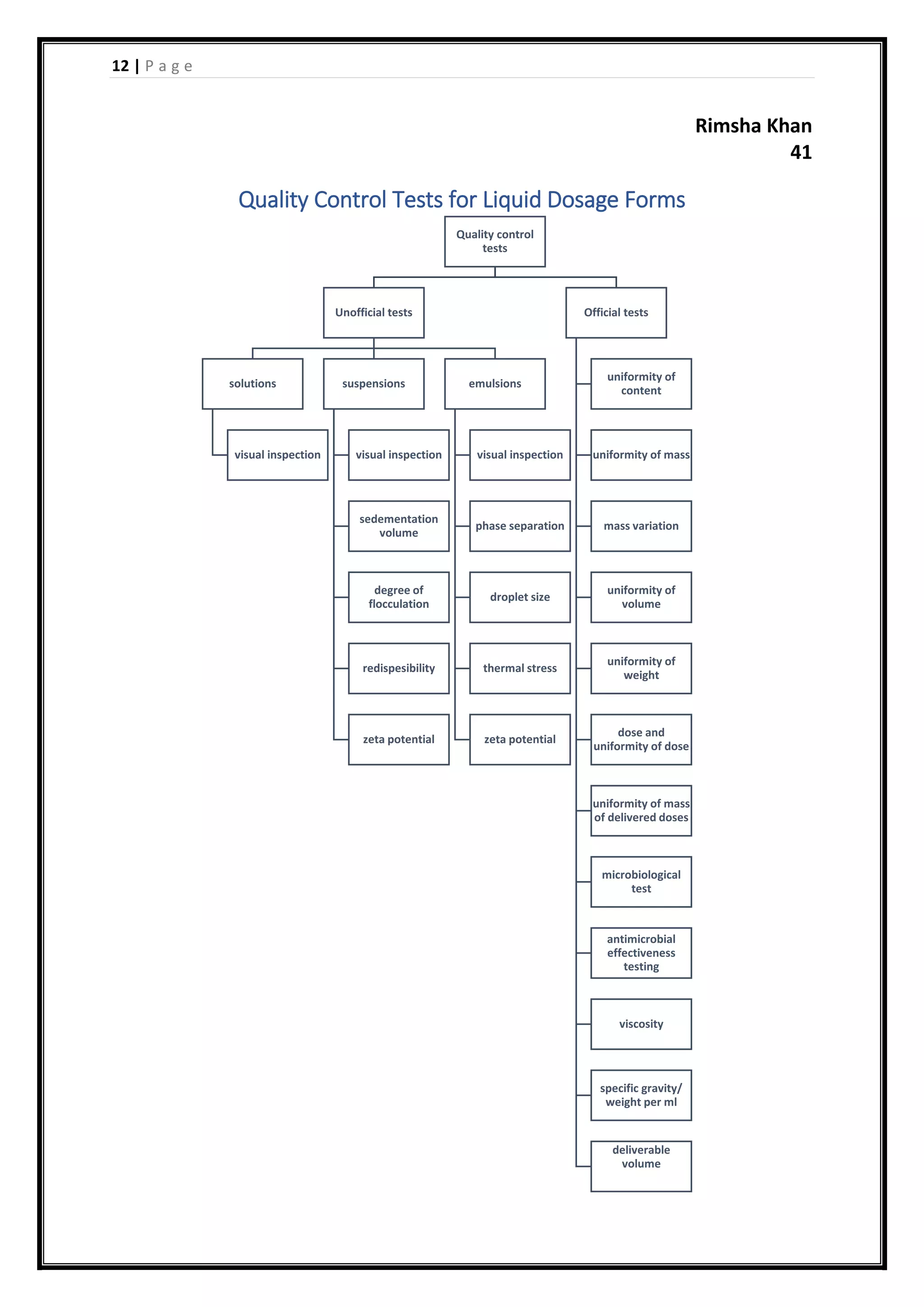
This document provides information on quality control tests for liquid dosage forms including oral solutions, suspensions, and emulsions. It discusses both unofficial tests like visual inspection, phase separation, and zeta potential, as well as official tests including uniformity of content, mass variation, specific gravity, and microbiological testing. The document serves as a guide for manufacturers to ensure liquid drugs meet specifications for identity, strength, quality, and purity.