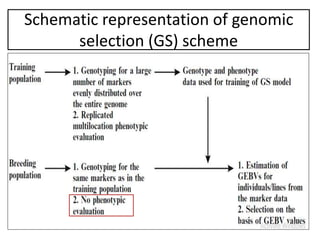
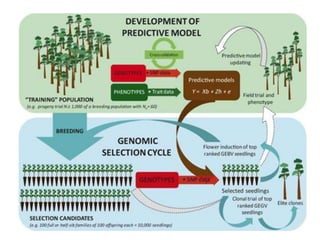
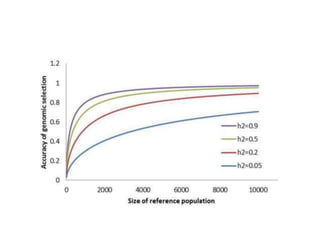
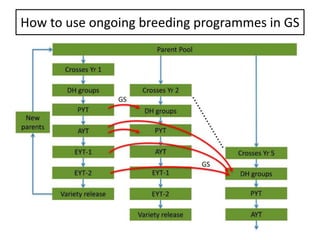
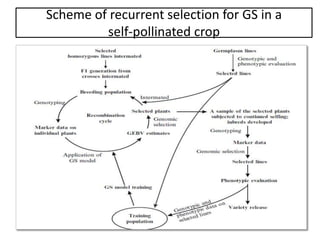
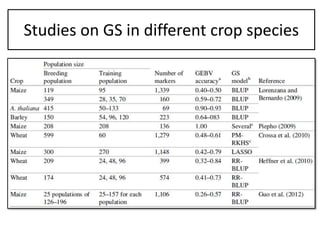
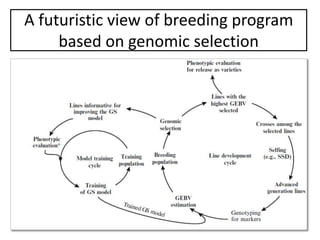
Genomic selection (GS), introduced by Meuwissen et al. in 2001, enhances marker-assisted selection by utilizing genome-wide marker data, enabling the prediction of genomic estimated breeding values (GEBVs) to select individuals in breeding populations. Compared to traditional methods, GS can more accurately predict breeding performance and is advantageous for traits with low heritability, thus improving efficiency in breeding programs. Despite its benefits, GS faces challenges like the need for extensive marker data and periodic model updates to account for changing genetic interactions.