Downloaded 64 times

























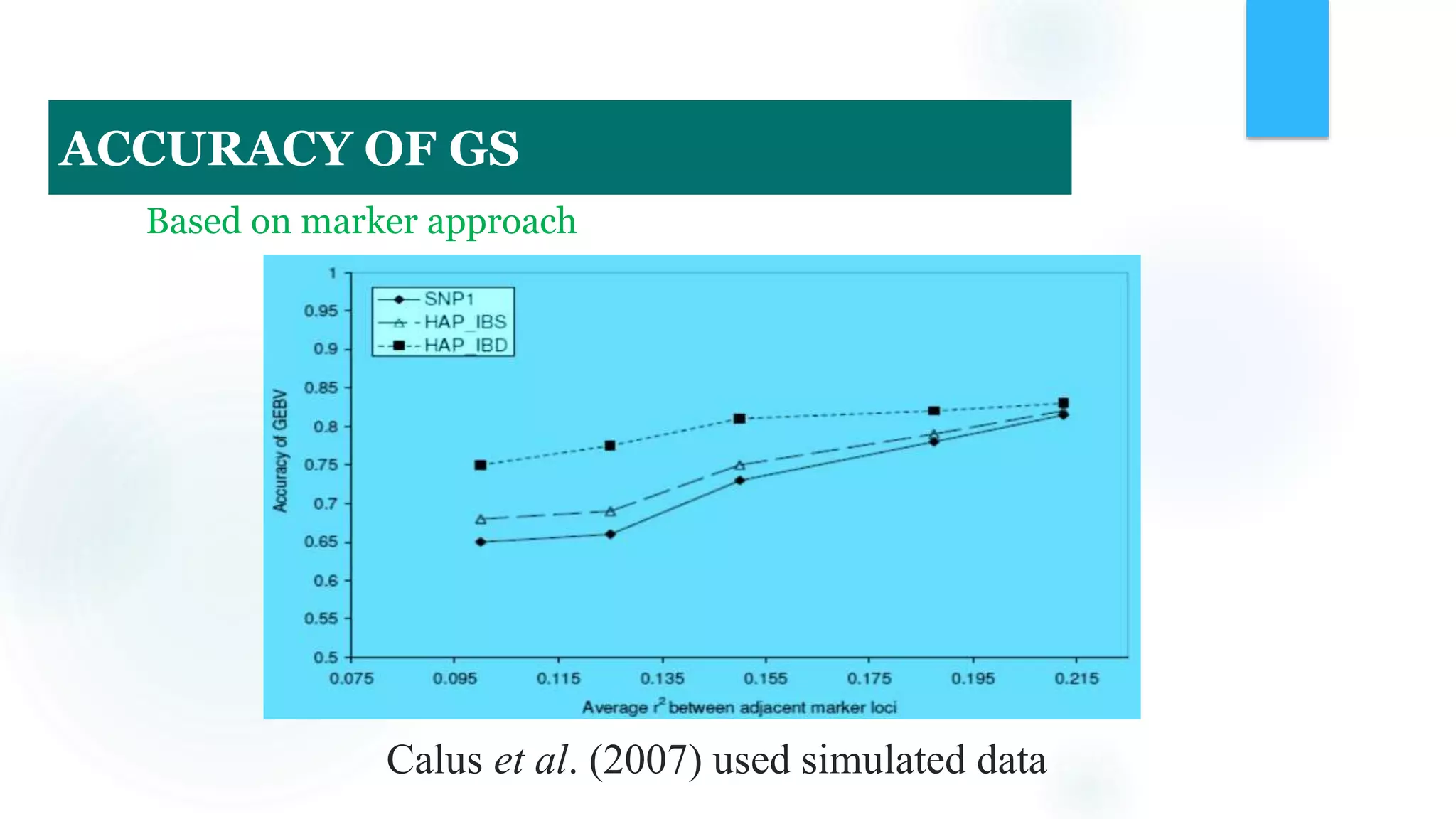


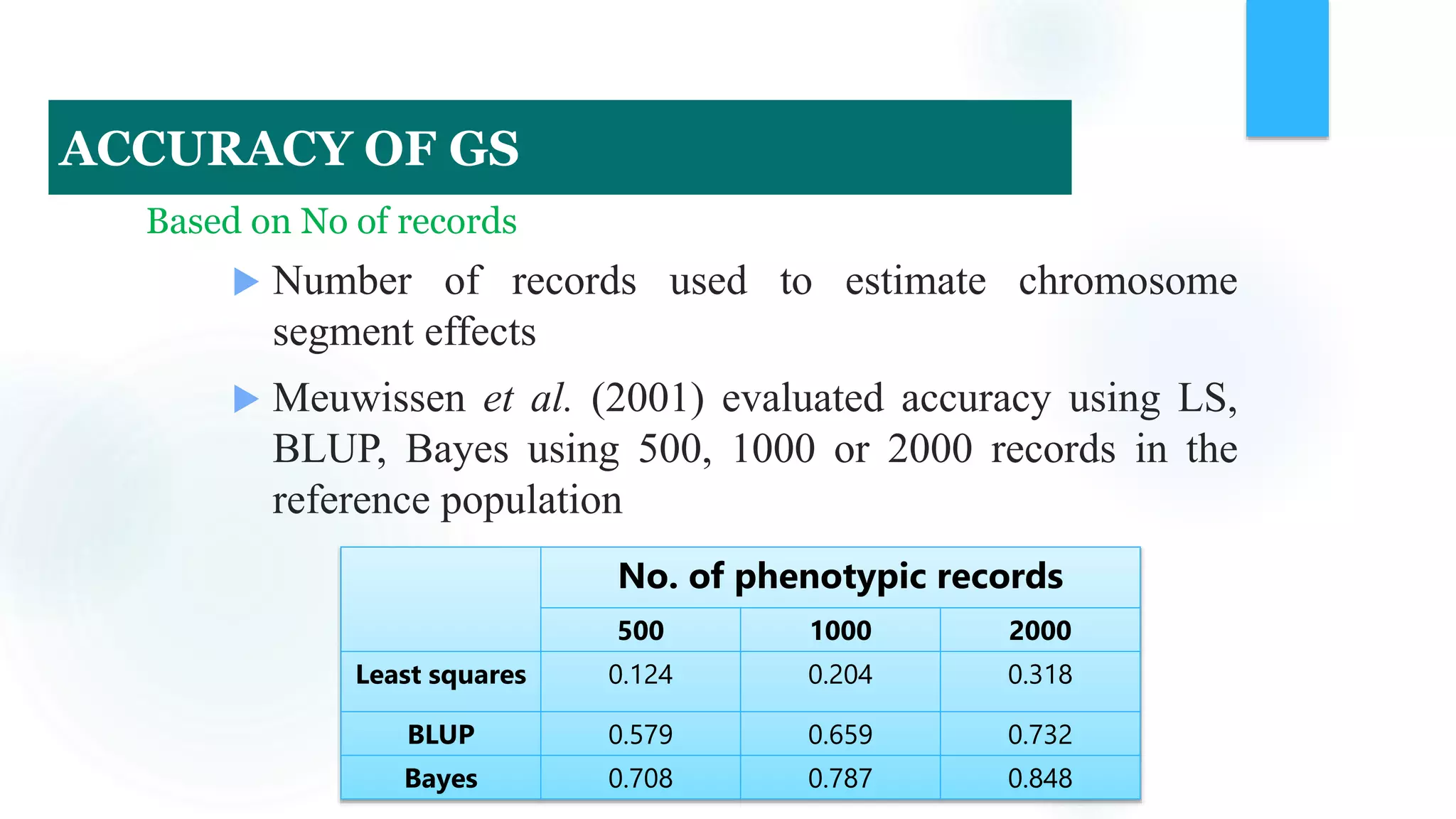
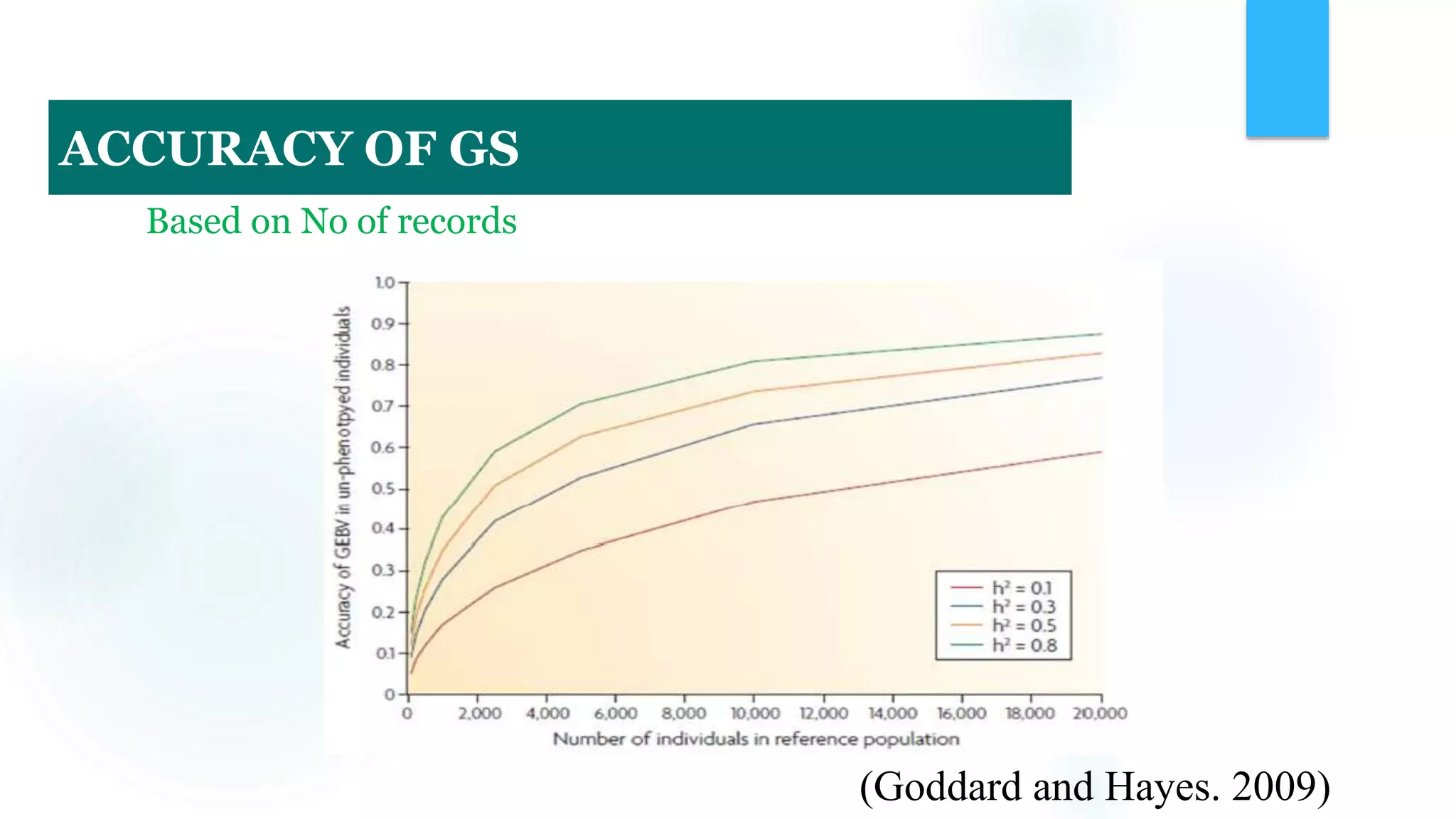
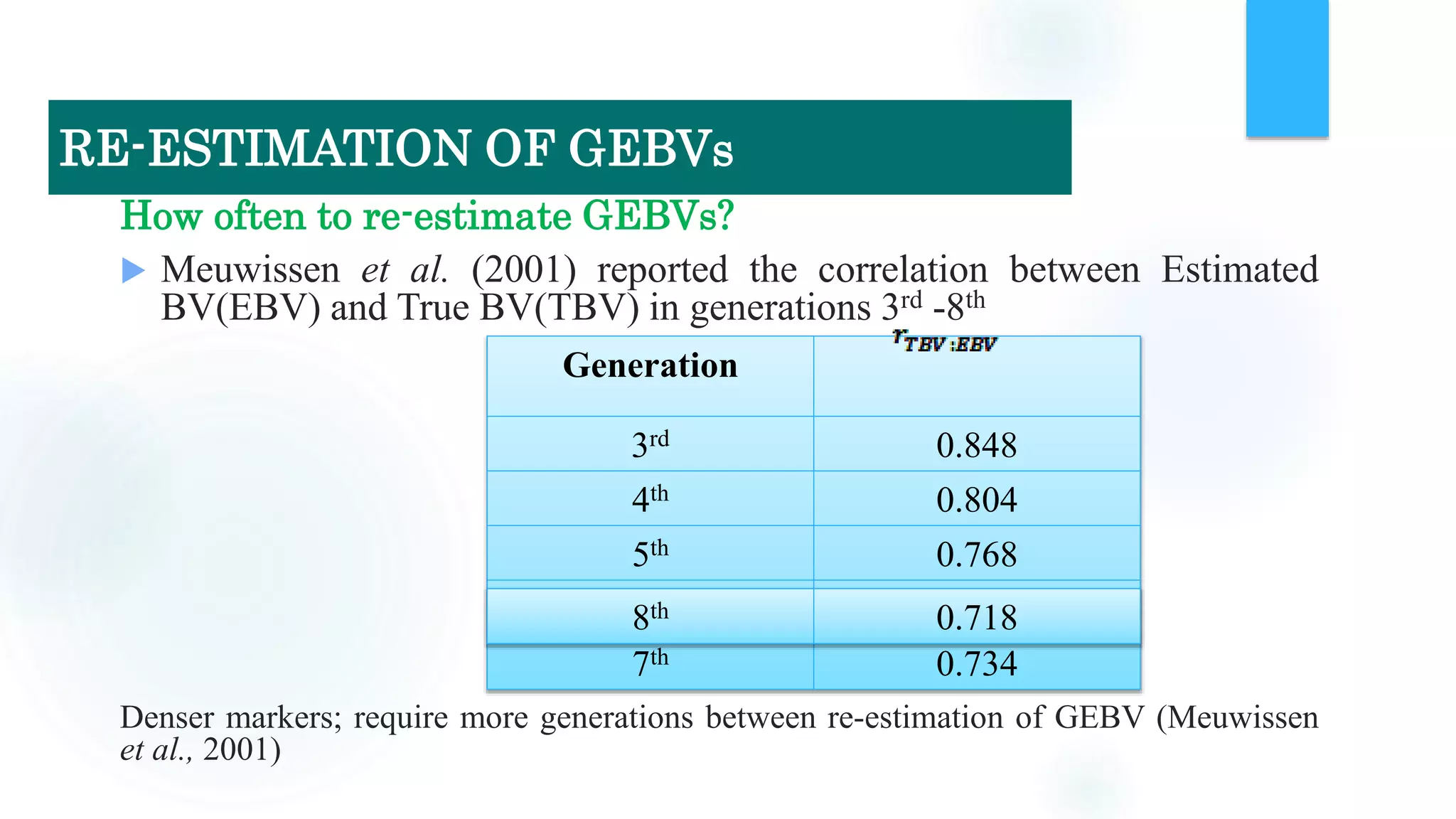





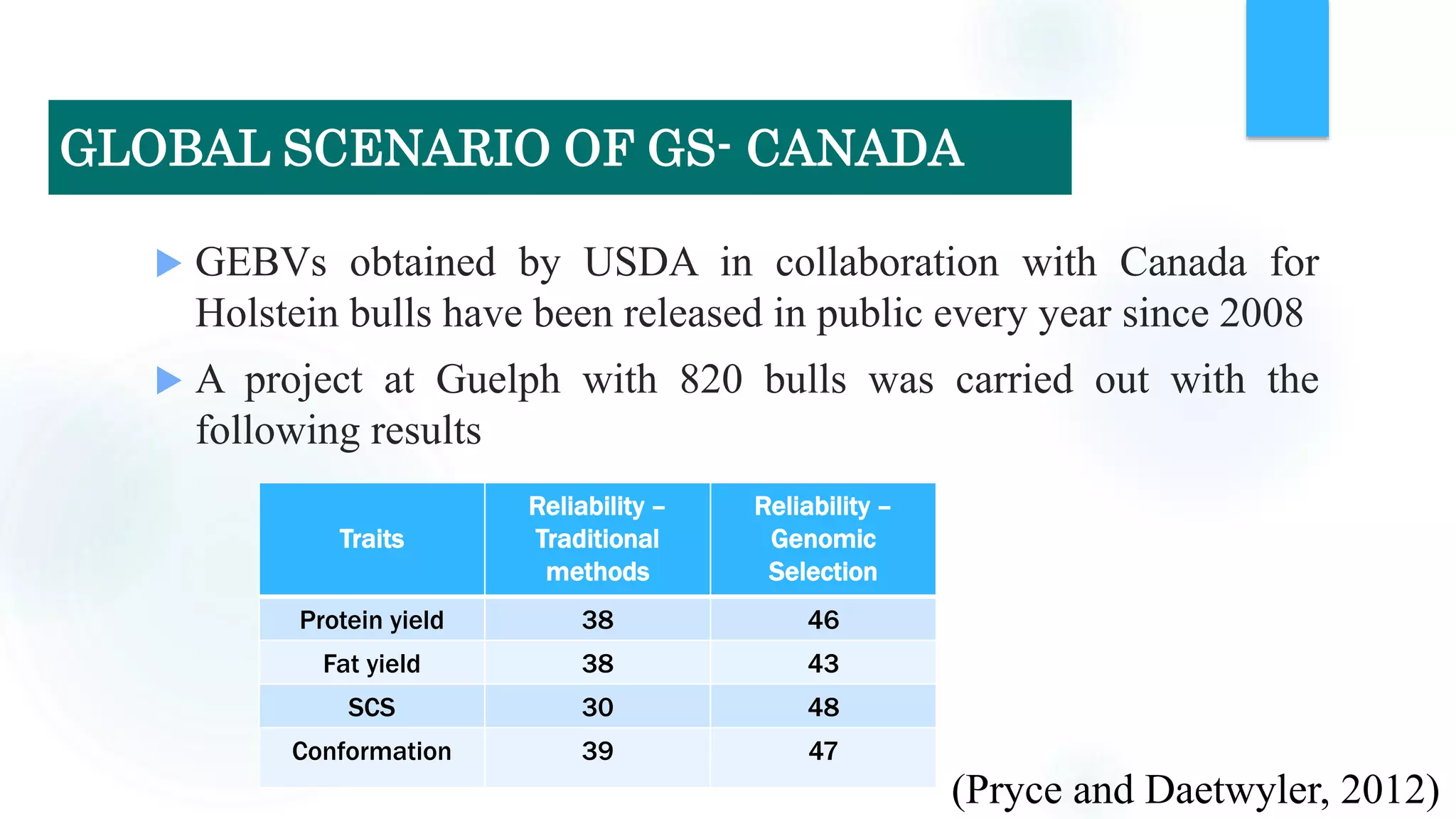
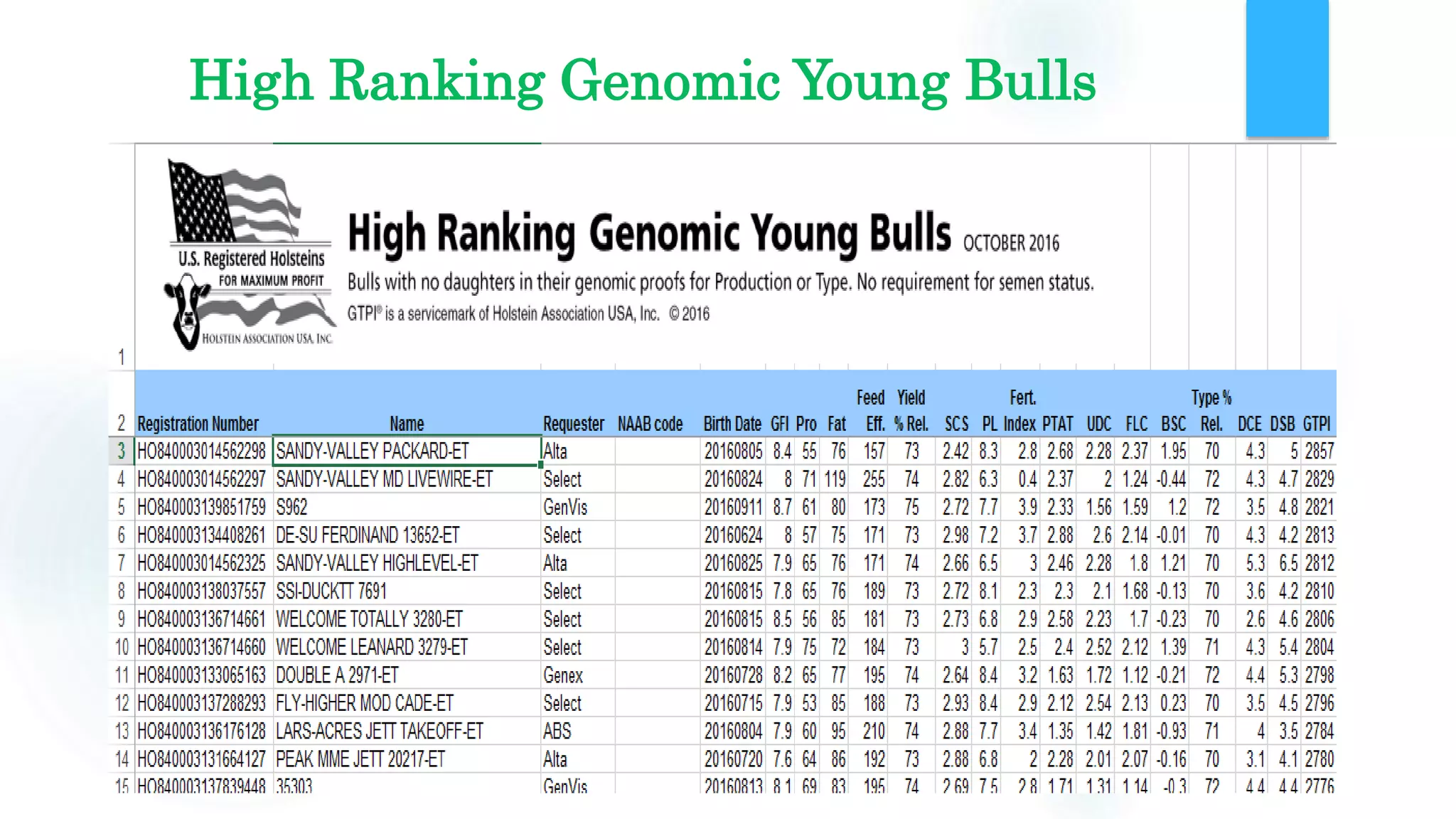






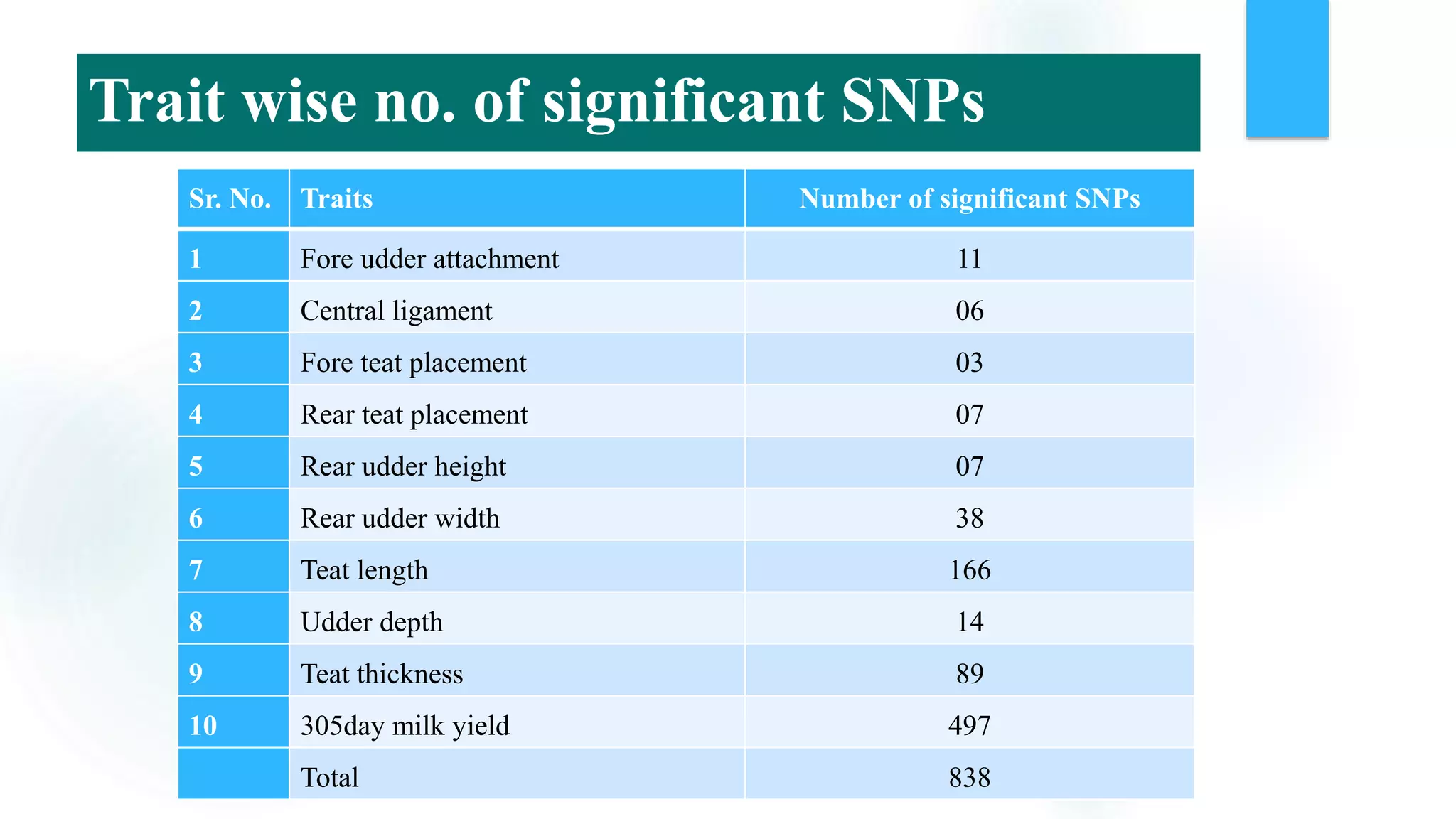
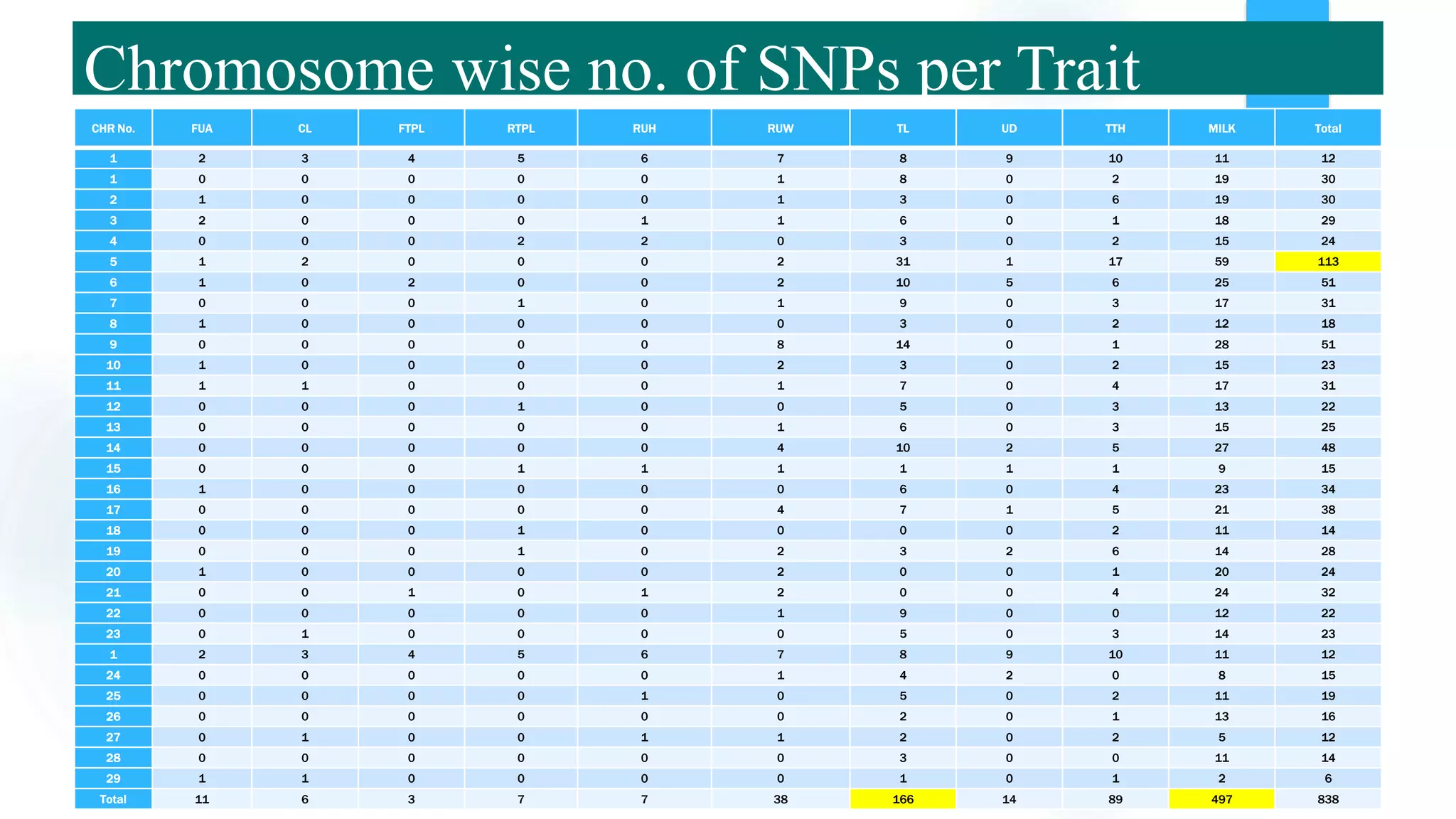
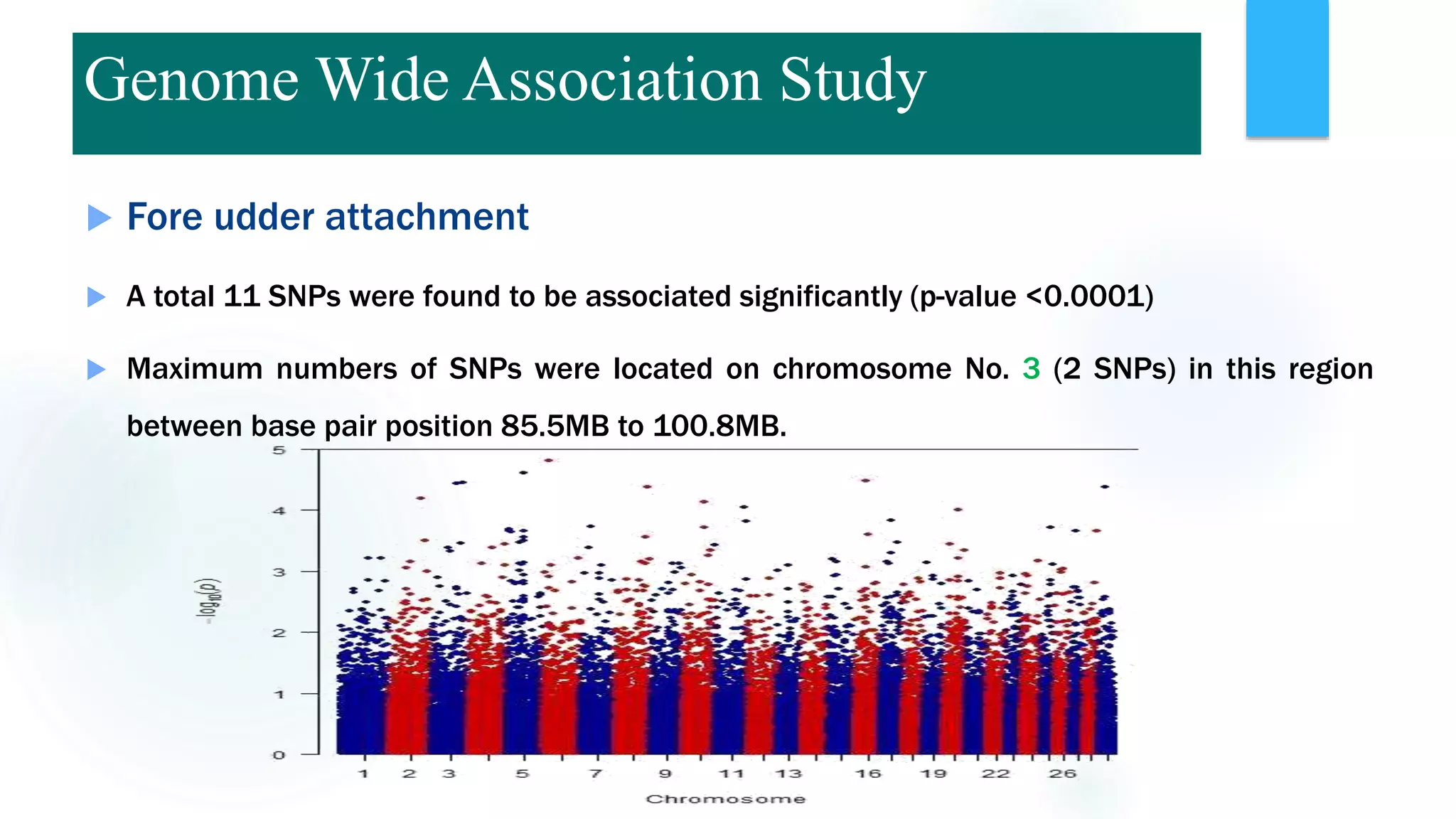
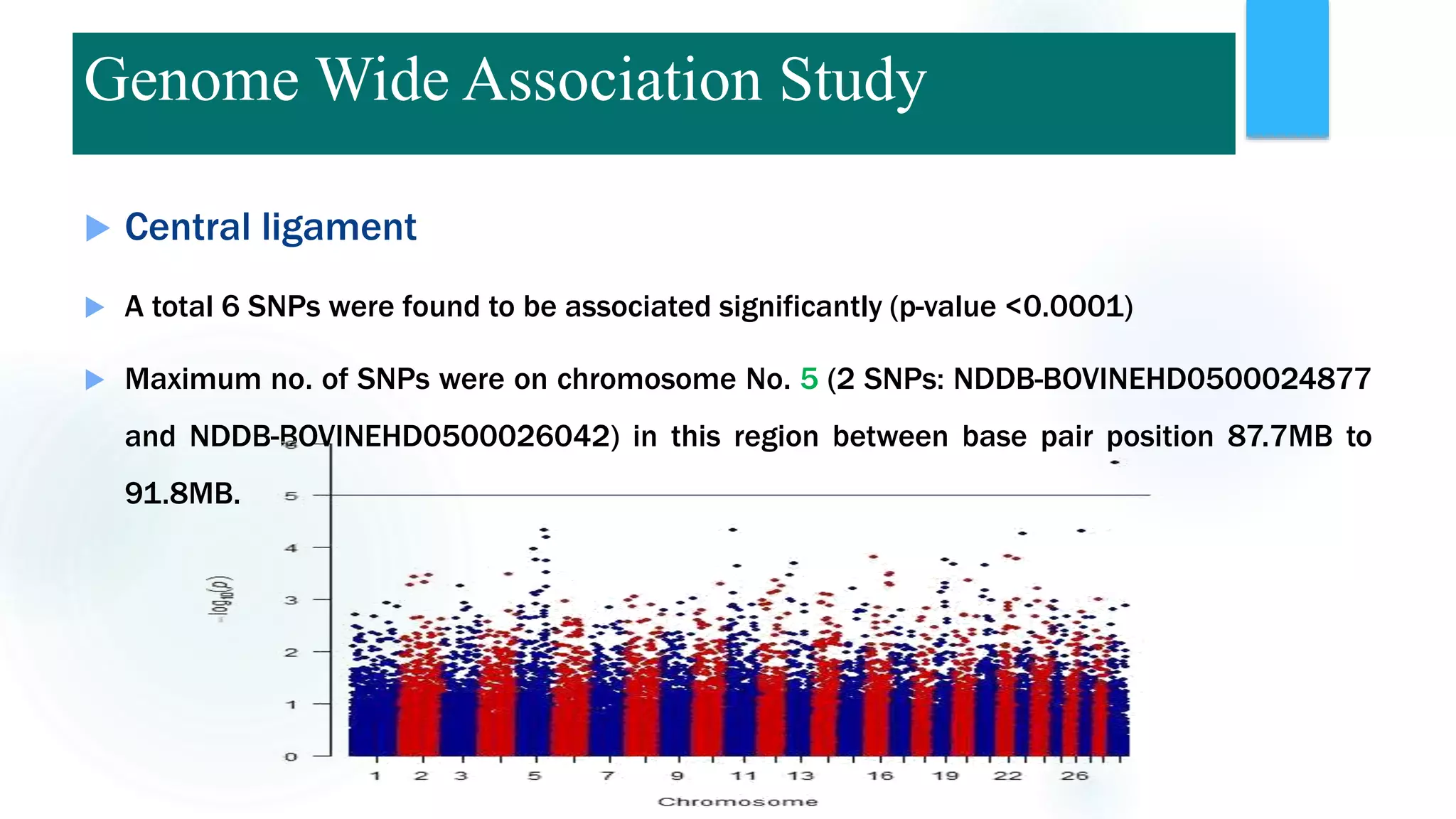
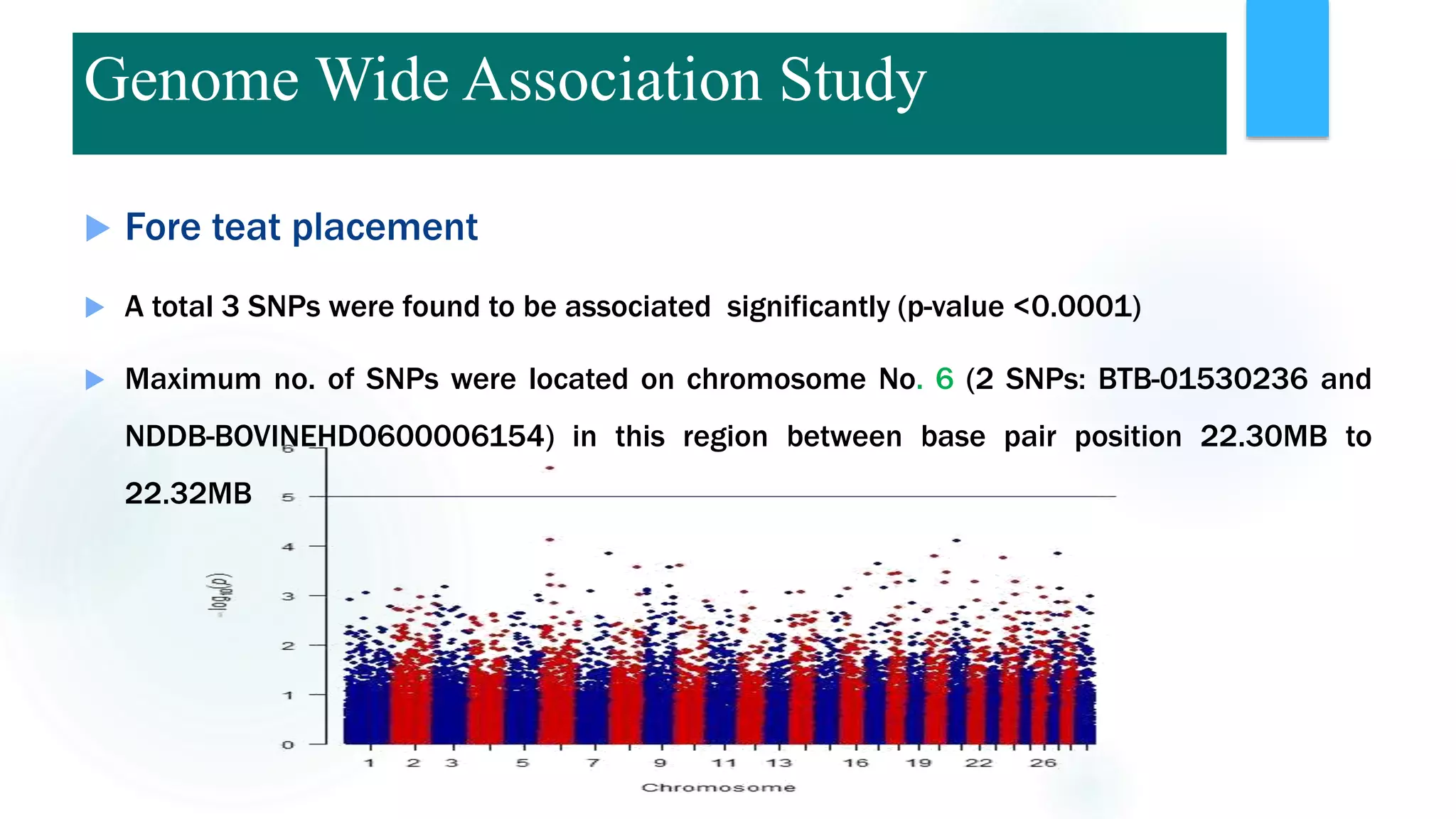
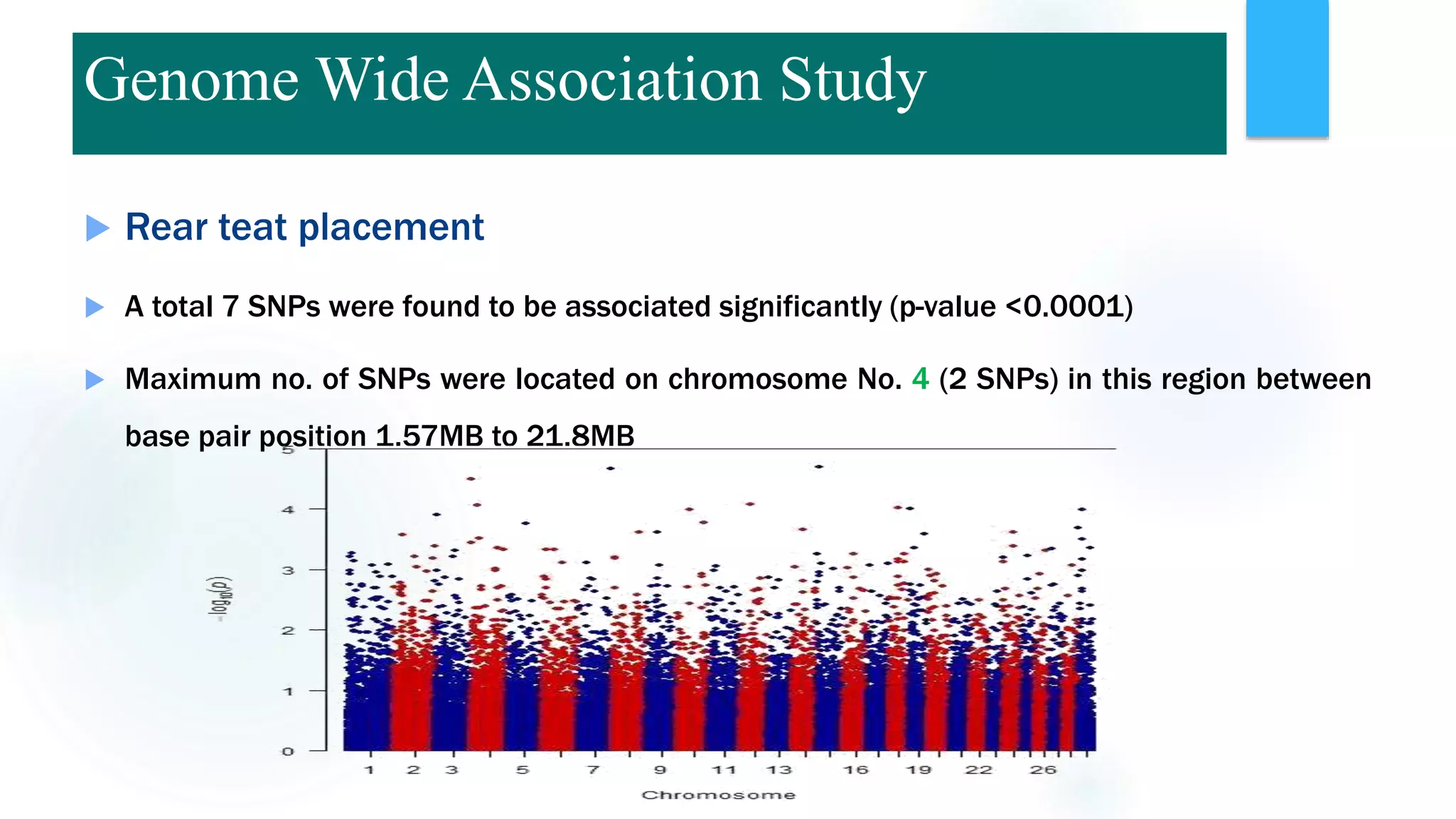
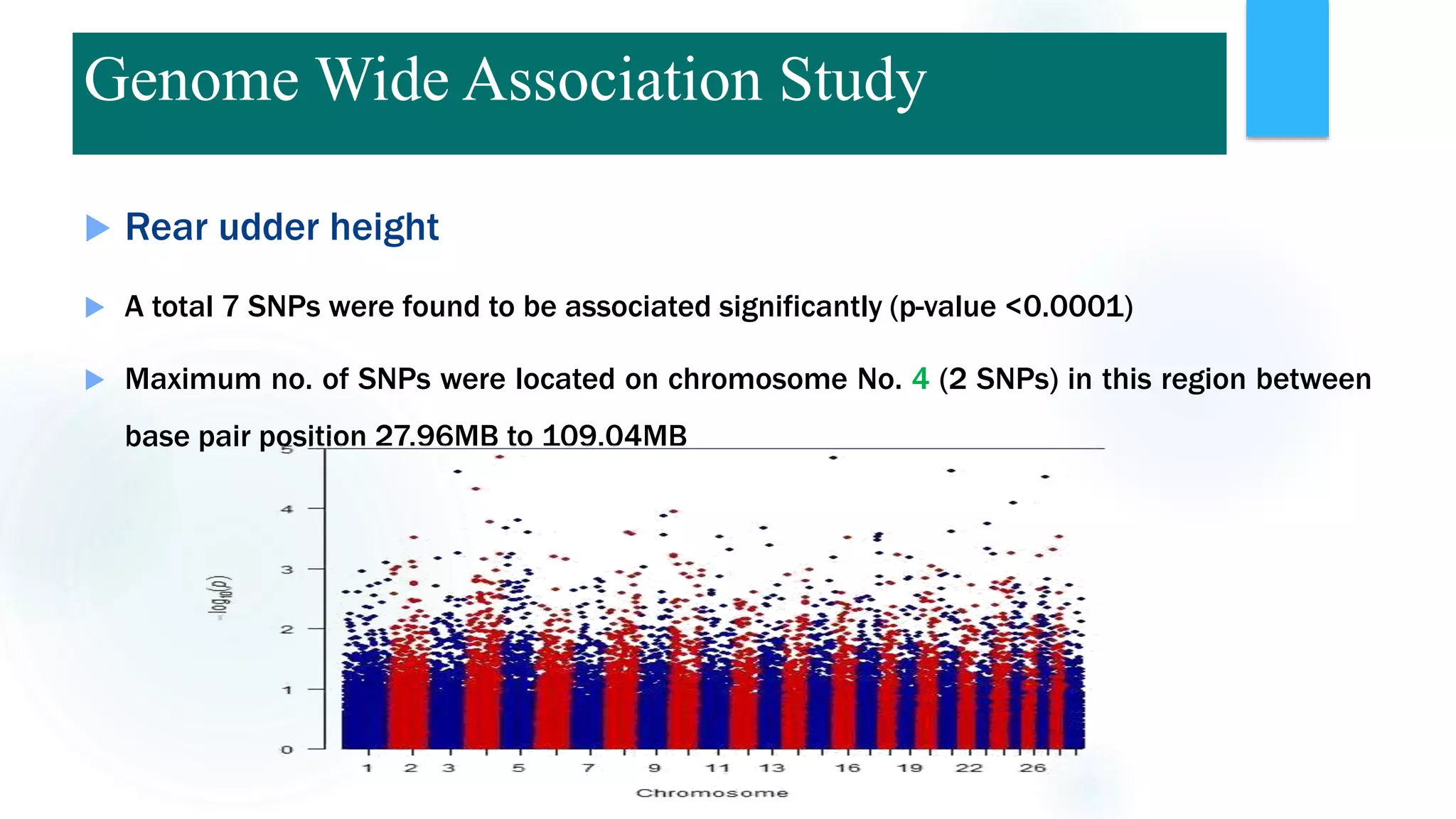
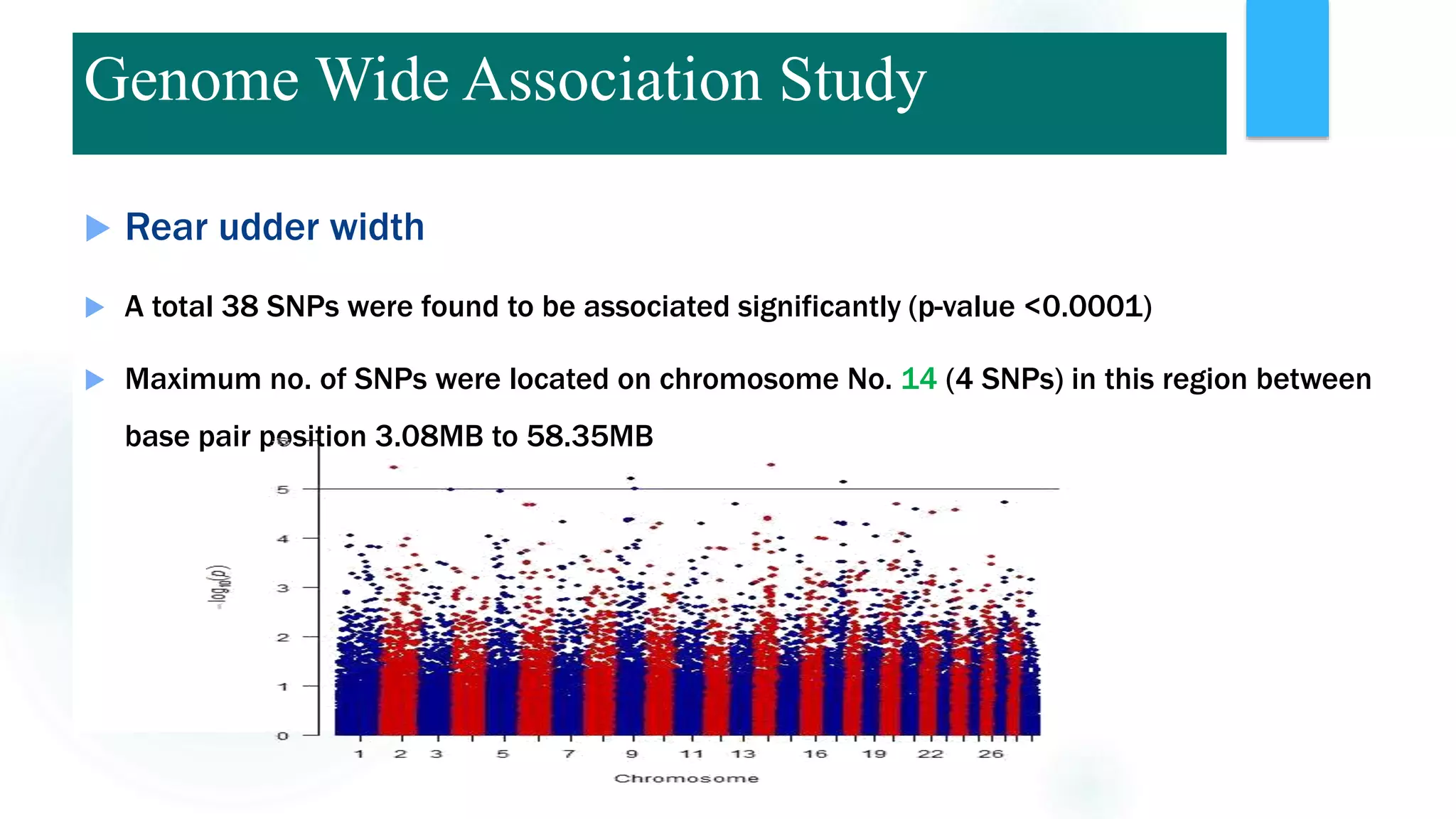
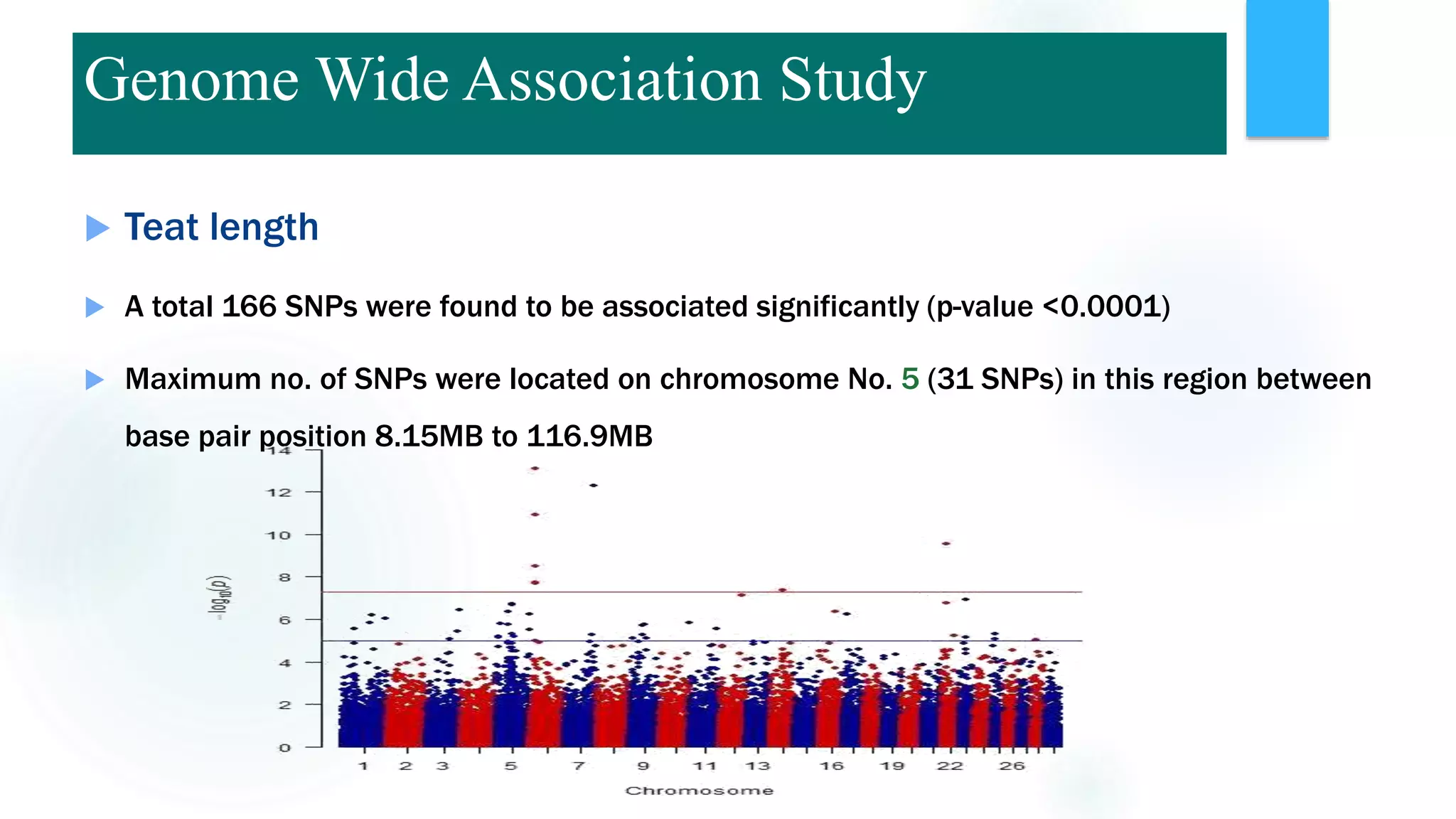
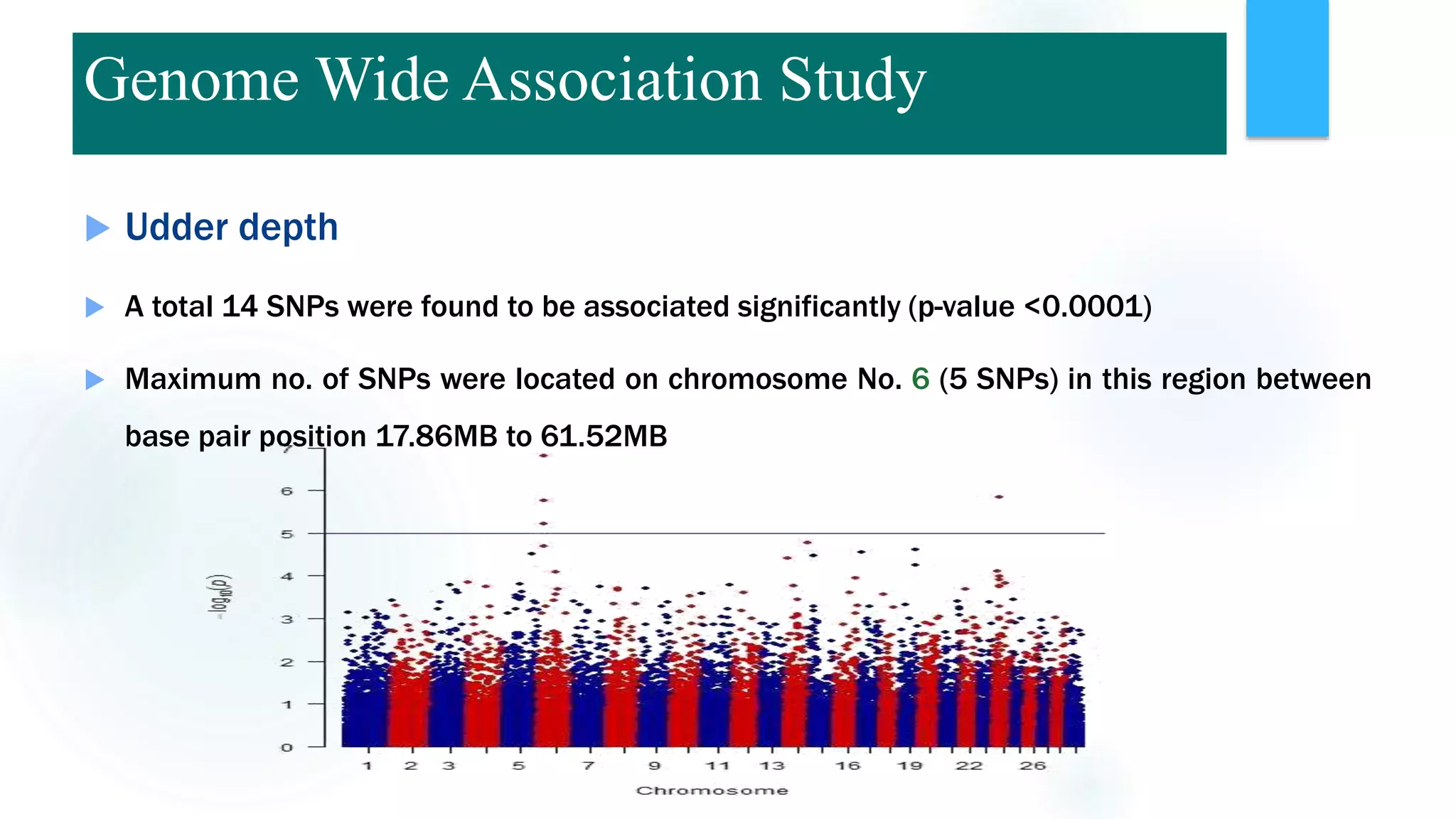
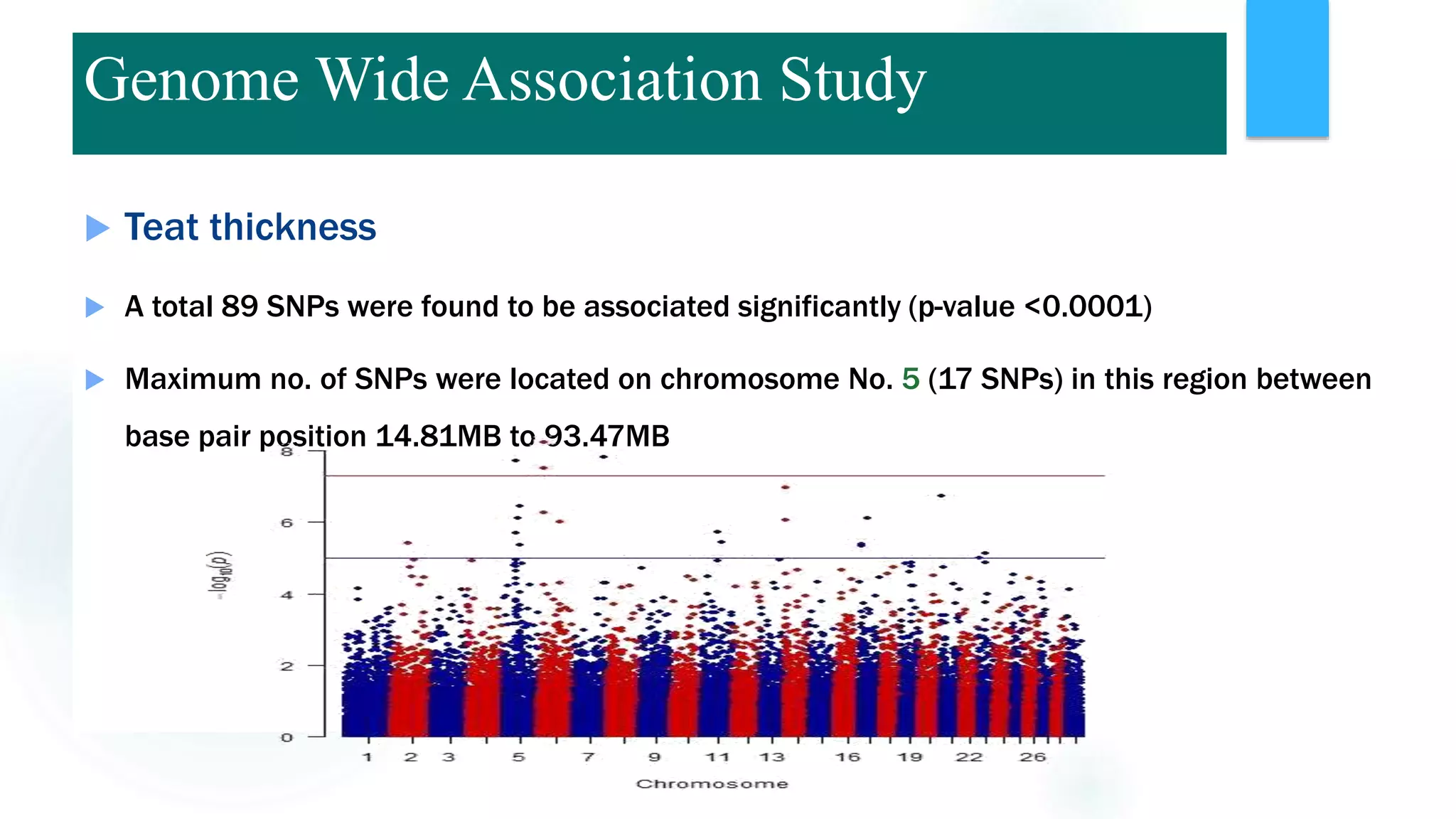
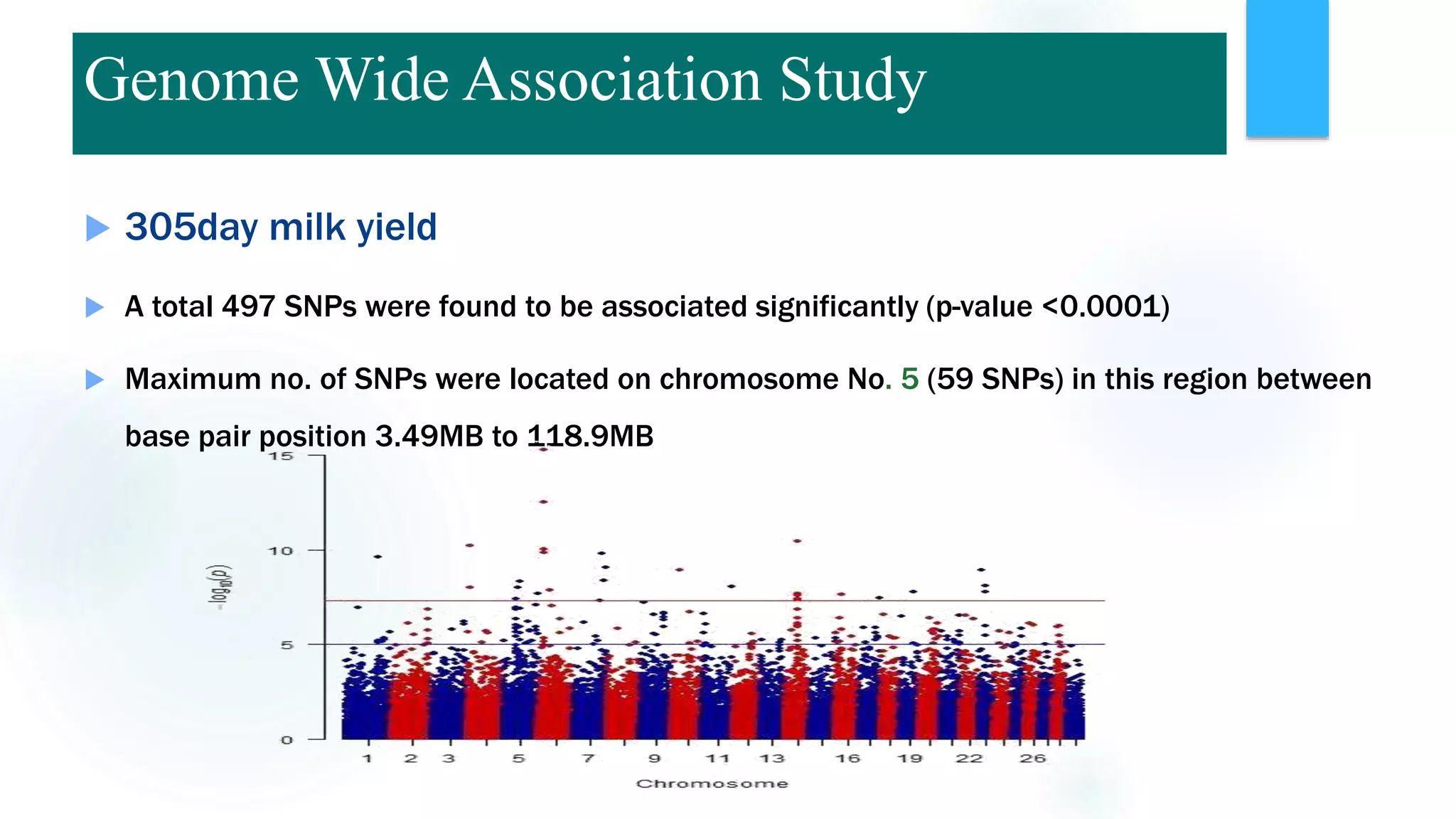



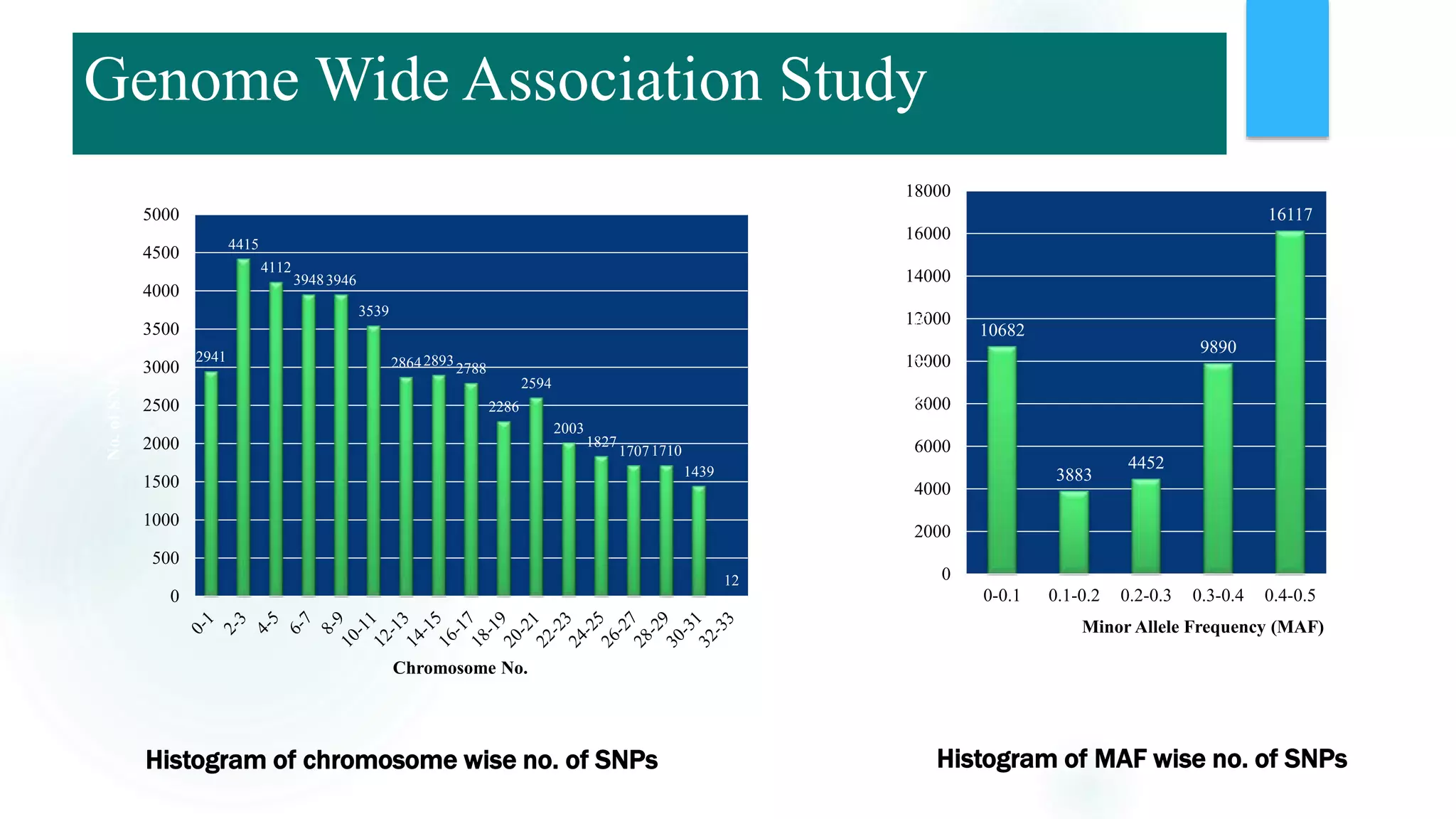
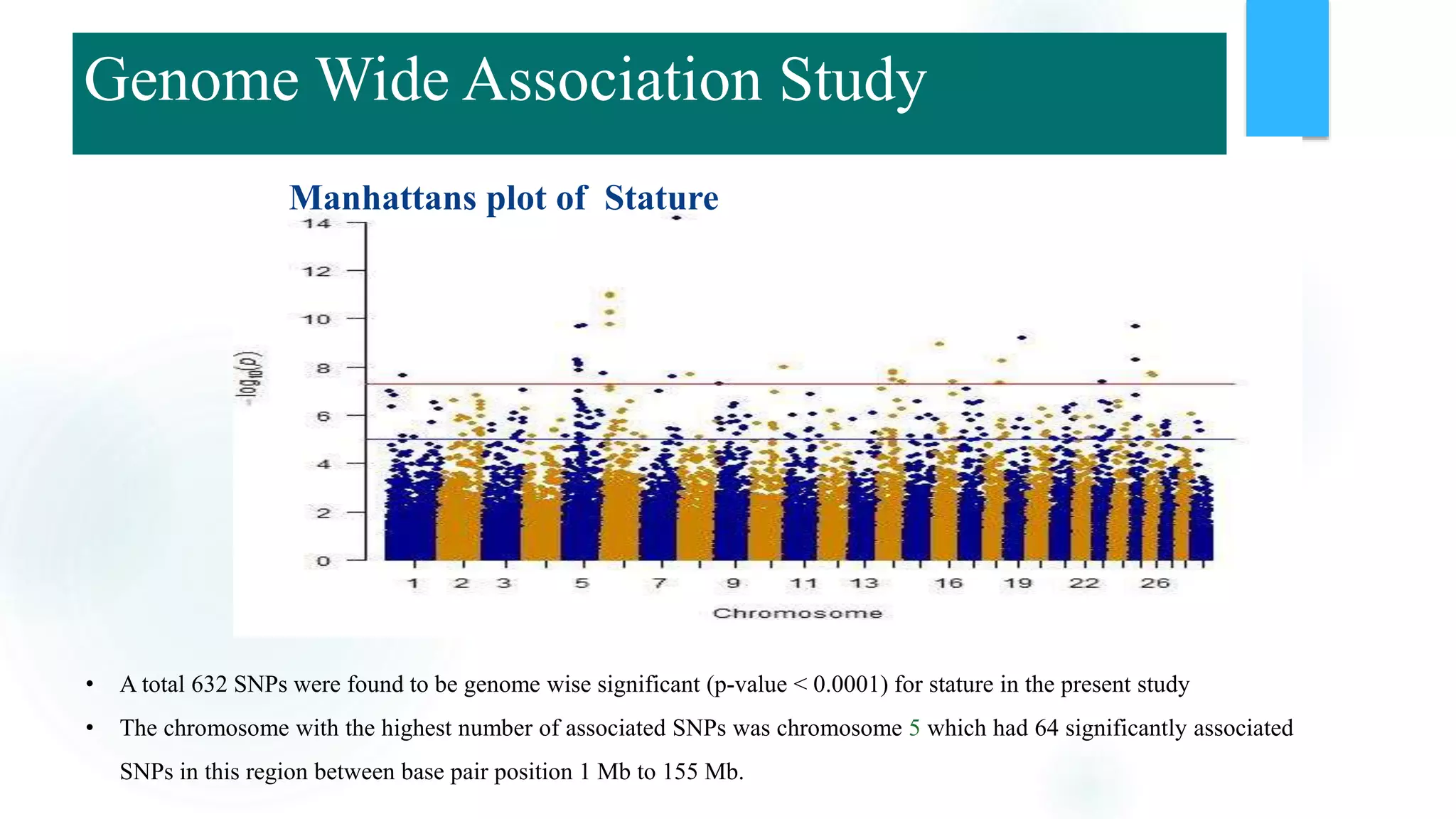
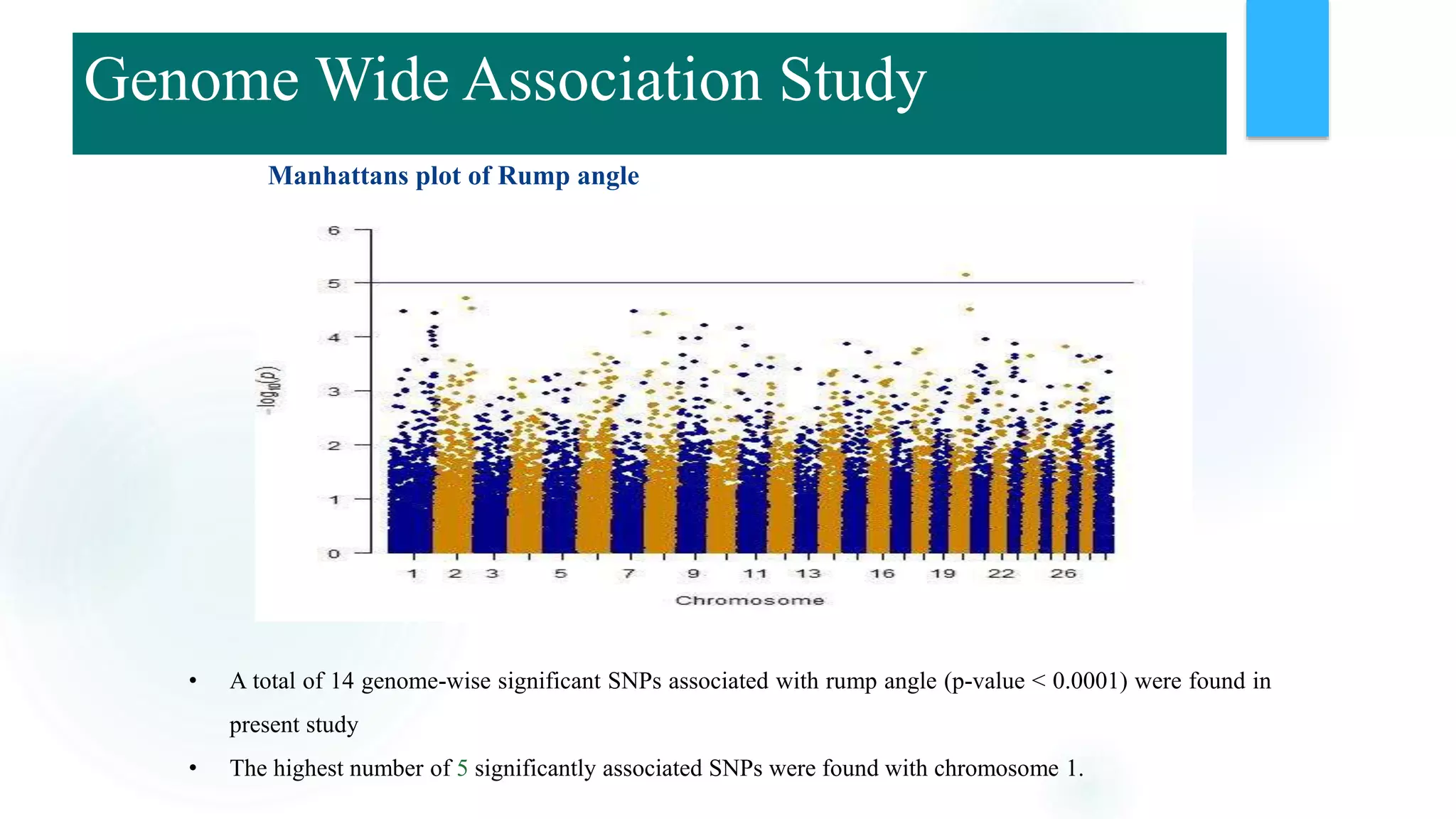
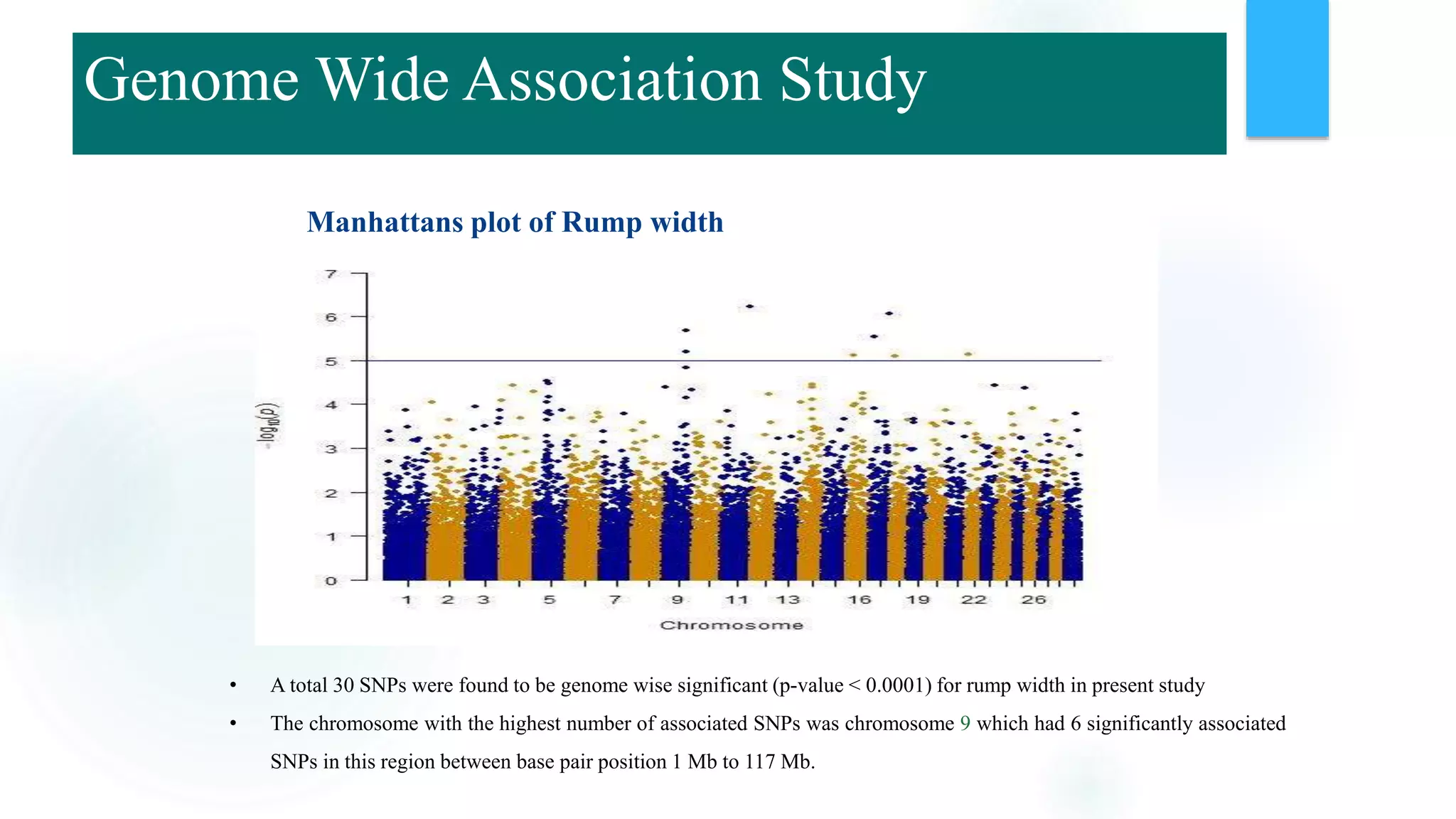
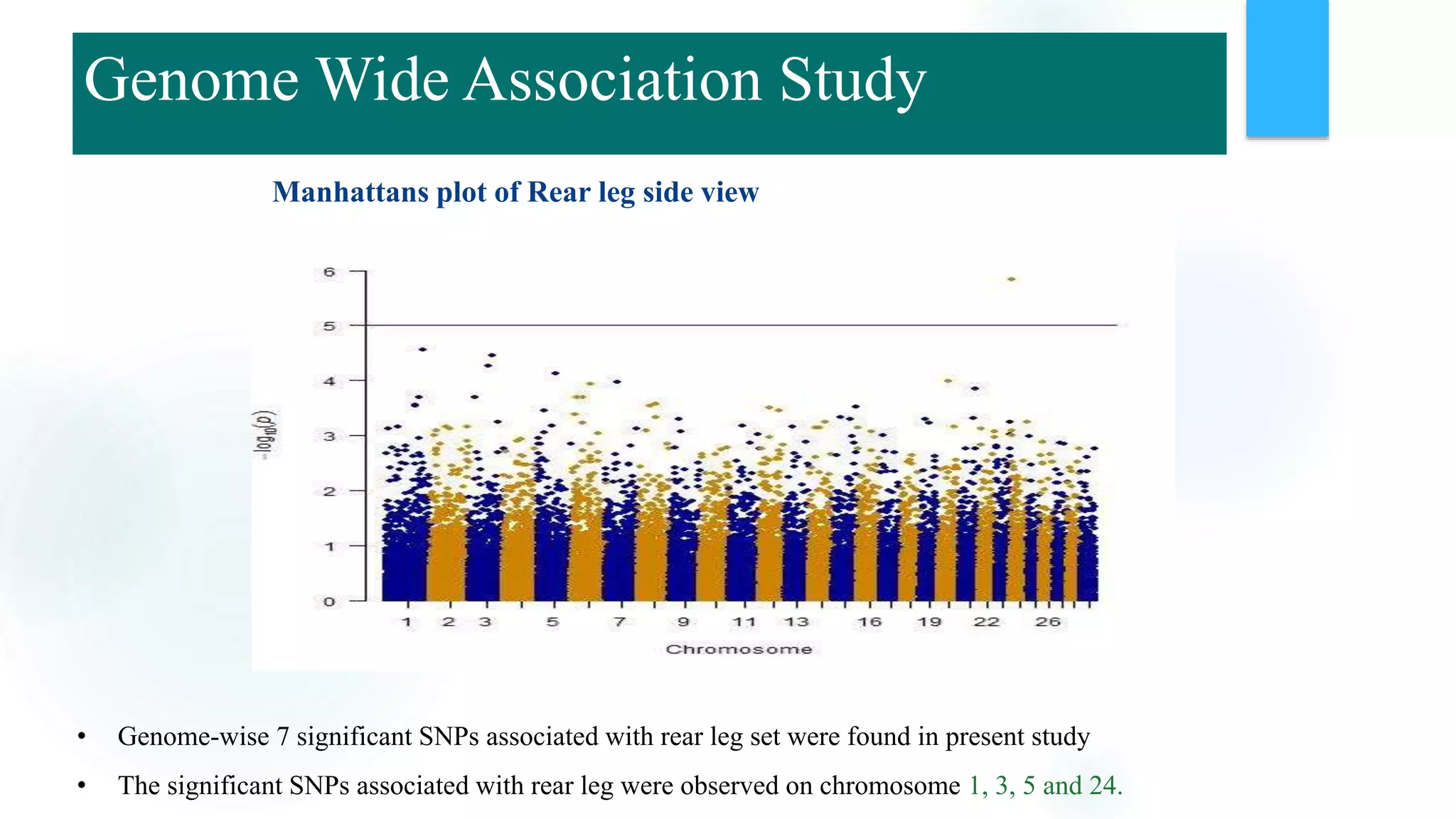
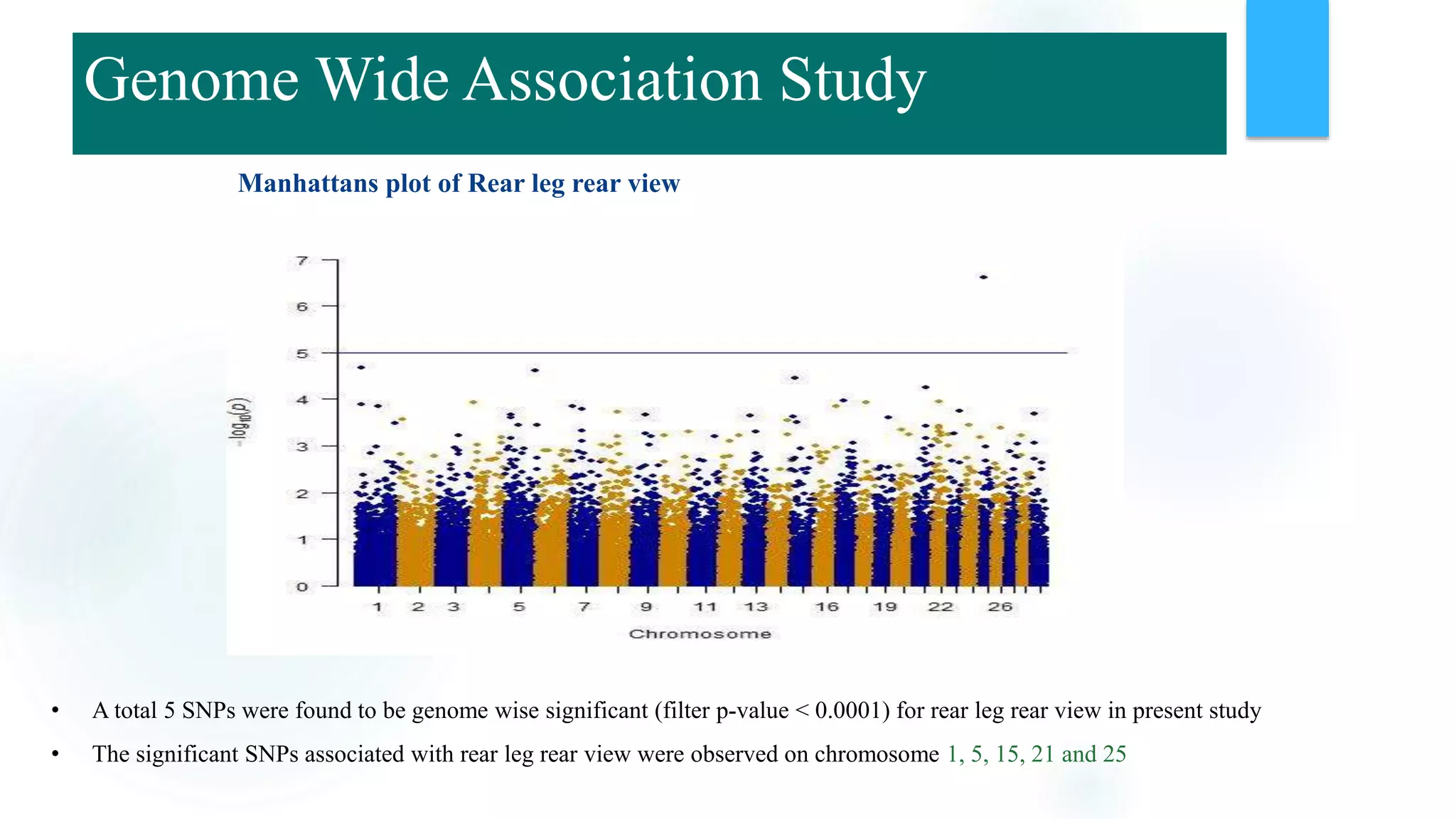
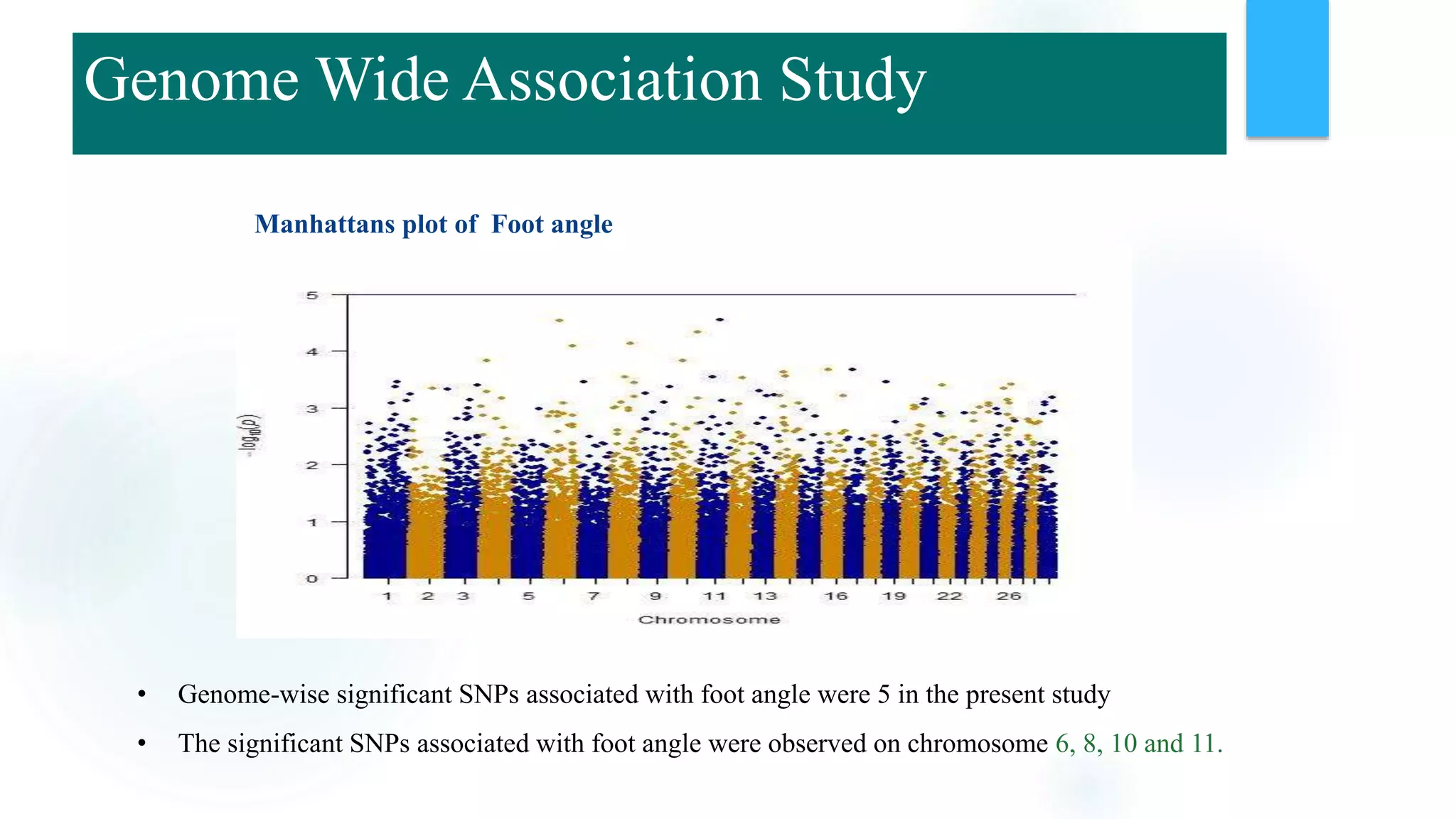
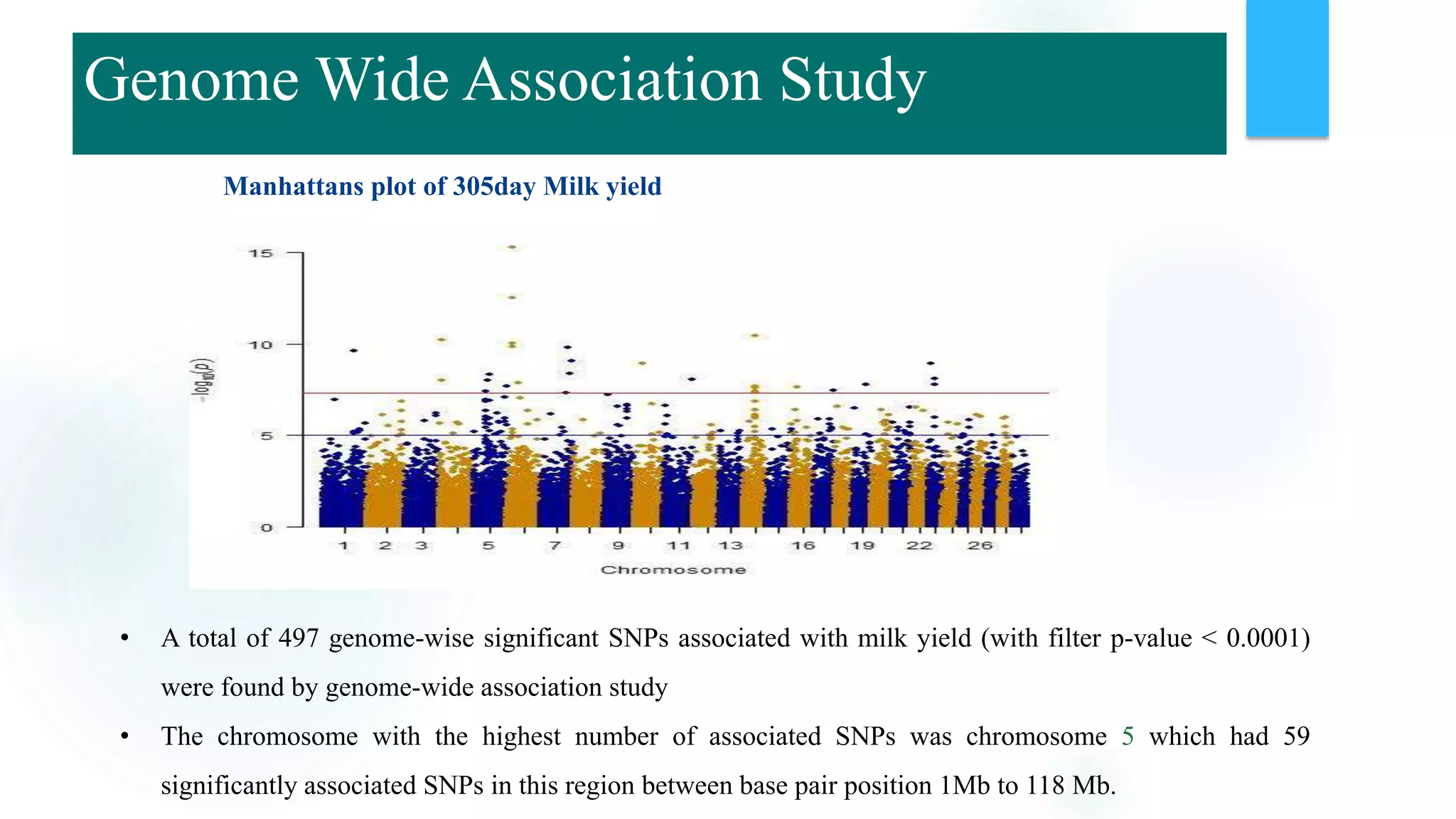






The document discusses the potential of genomic selection in indigenous cattle breeds, highlighting its methodology, advantages, limitations, and impact on animal breeding. It details the shift from traditional breeding methods to modern genomic approaches utilizing SNP markers and emphasizes the need for accurate phenotypic records. The findings suggest significant improvements in breeding efficiency, genetic gain, and selection accuracy, particularly for traits that are typically challenging or costly to measure.



































































