Download to read offline

























![• Drabkin’s reagent (0.2 g potassium ferricyanide -K3[Fe(CN)6], 1.0g sodium carbonate, 0.5
ml of acetoncyanhydrine
CH3 C CH3
CN
OH
• Hemoglobin standard solution (140 g/L)
• Sample of patient’s serum
• Blood (Sahli) pipettes, 0.02 ml
• Test tubes
• Colorimeter
Procedure:
• Take three test tubes and mark them B (blank), R (reference) and P (patient)
respectively.
• Prepare solution according to the table:
Table Preparing of the working solution for determination of blood hemoglobin
№ Reagents B R P
1. Drabkin’s reagent 5.0 ml - 5.0 ml
2. Hemoglobin standard
solution
- 5.0 ml -
3. Serum - - 0.02 ml
Let the test tube stand
at room temperature
for 10 minutes
• Mix the contents of each test tube.
• Read the optical density (O.D.) of solutions from P and R test tubes against blank
solution at 490-540 nm transmissions (green filter) in a colorimeter using cuvette with
1.0 cm thickness of medium through light passes.
Calculation
Calculate the concentration of blood hemoglobin using the following formula:
Blood hemoglobin
concentration =
(g/L)
O.D.(P)
O.D.(R)
140 g/L
Where,
34](https://image.slidesharecdn.com/fibrousproteinglobularprotein-190312112231/85/Fibrous-protein-Globular-protein-34-320.jpg)





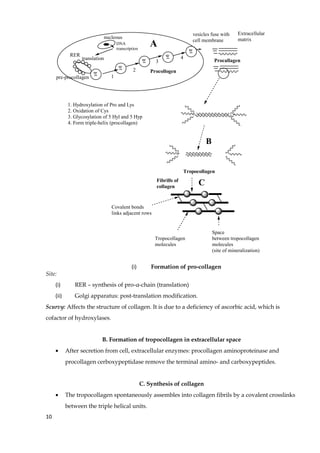
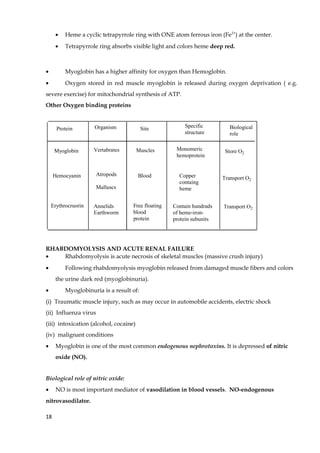
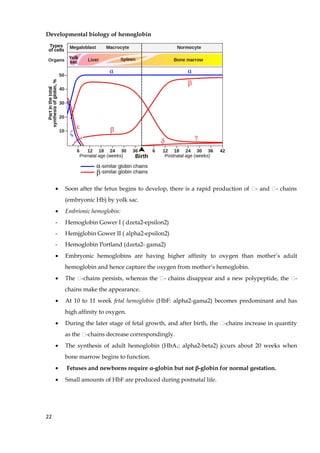
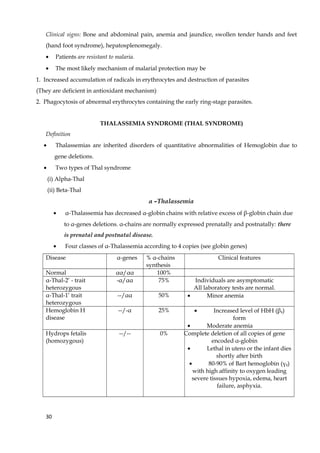
1. The document discusses fibrous and globular proteins, focusing on collagen, elastin, myoglobin, and hemoglobin. It provides details on the structure, function, and biosynthesis of these proteins. 2. Collagen is the main fibrous protein in the body. It has a characteristic triple helical structure that gives tissues strength and elasticity. Defects in collagen can lead to conditions like Ehlers-Danlos syndrome and osteogenesis imperfecta. 3. Elastin provides elasticity to tissues like lungs, blood vessels, and skin. It forms cross-links that allow these tissues to stretch and return to their original shape. Mutations can result in Morfan syndrome.























































































