Download as PDF, PPTX

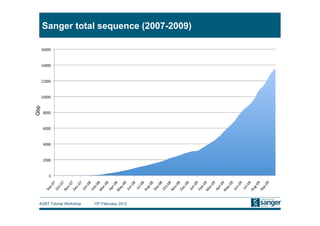
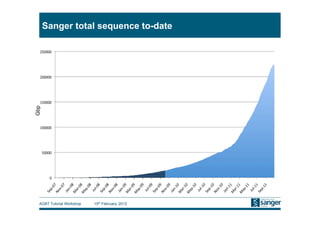


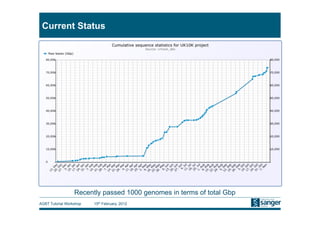

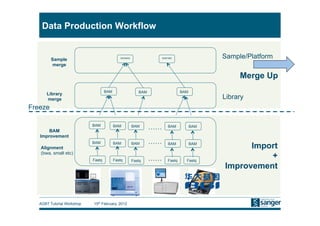
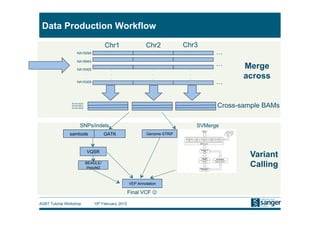

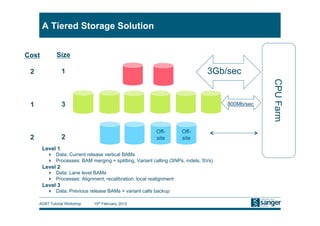
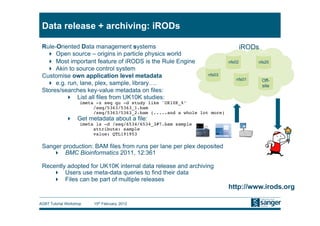

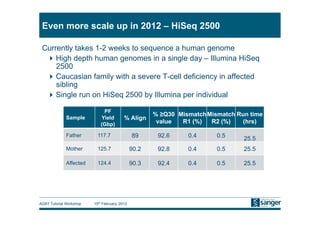
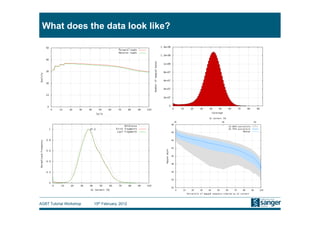

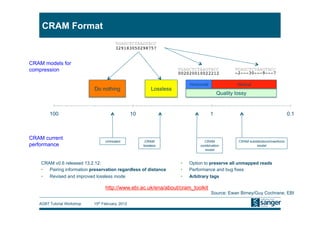


This document discusses the challenges of large scale DNA resequencing projects at the Wellcome Trust Sanger Institute. It describes their work on several major projects that have generated over 100 terabases of sequencing data, including the 1000 Genomes Project and UK10K. It outlines their data processing pipeline and strategies for managing the large amount of data through tiered storage and automated compute workflows. Finally, it notes that new high throughput sequencers like the Illumina HiSeq 2500 will allow generating a human genome in a single day, further increasing the scale of data production.






















































![5G Explained! A High Level Overview [Introduction]](https://cdn.slidesharecdn.com/ss_thumbnails/5gexplainedahighleveloverview-260119165306-cc137a3e-thumbnail.jpg?width=640&height=640&fit=bounds)
















