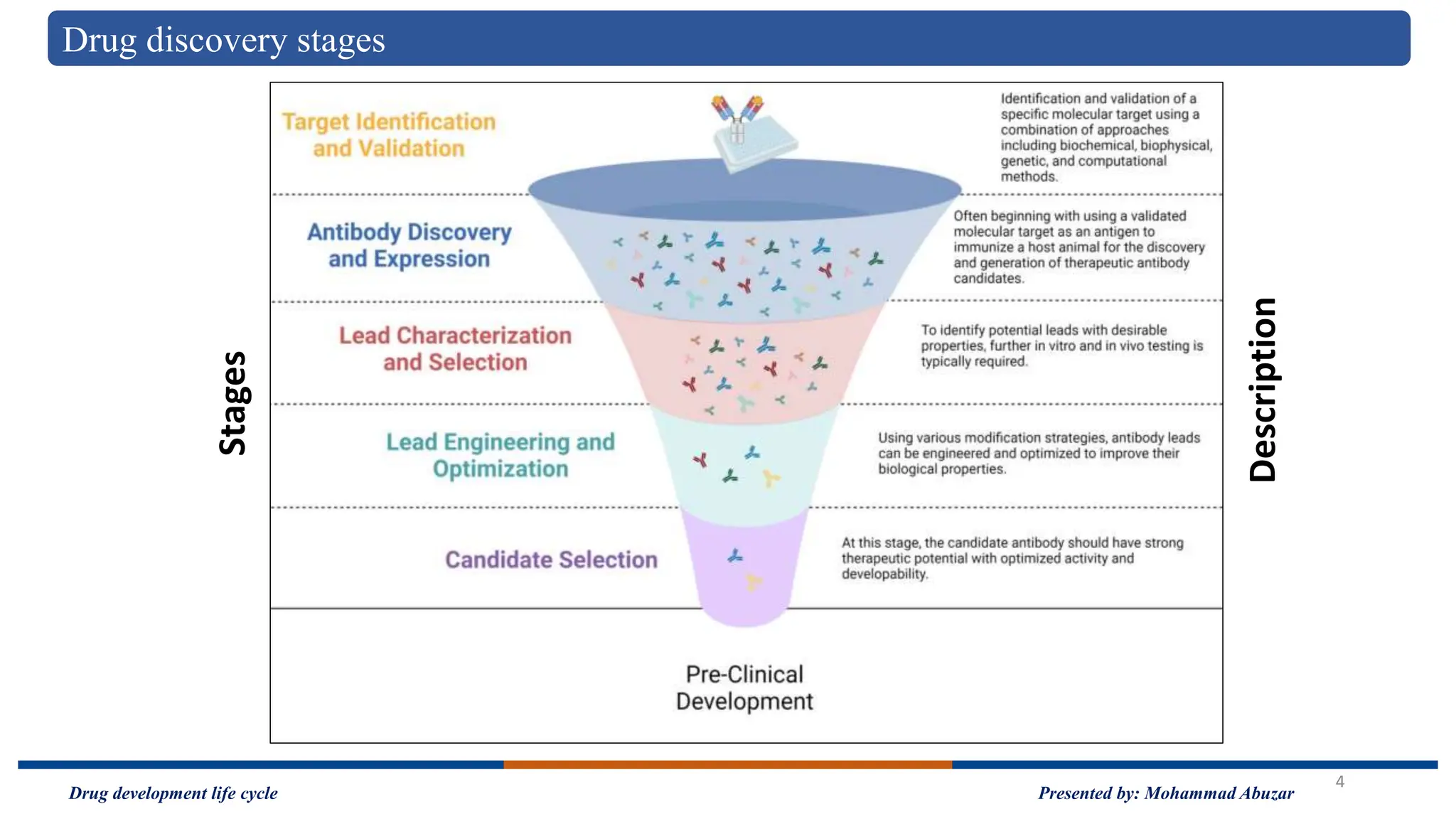
The document outlines the drug development life cycle, detailing the extensive time (10-15 years) and financial investment (approximately $800 million to $1 billion) required to bring a new medicine to market. It discusses various stages, including drug discovery, the investigational new drug application process, and the new drug application, as well as the differences between branded and generic drugs. Key concepts like side effects, adverse effects, biologics, and relevant legislation such as the Hatch-Waxman Act and the Biologics Price Competition and Innovation Act are also explored.