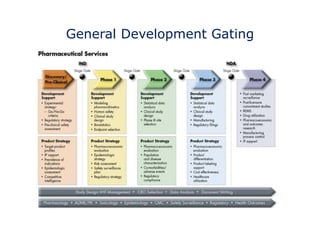
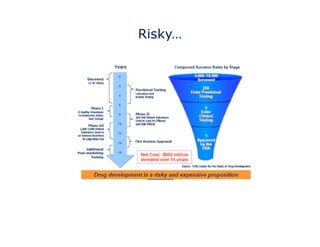
The document discusses the complex and unpredictable nature of the FDA drug approval process. While the steps of drug development may seem formulaic, including discovery, preclinical testing, and clinical trials, success is not guaranteed as programs face many risks and intangible factors. Understanding these challenges is important for mitigating risks and strategizing development approaches. The FDA approval process aims to ensure new drugs are safe and effective for patients.














![So what’s a drug ?
Drug describes [any] articles intended for use in
the diagnosis, cure, mitigation, treatment, or
prevention of disease in man or other animals.
15](https://image.slidesharecdn.com/fdaapplicationsinanutshell-141021085414-conversion-gate01/85/FDA-applications-in-a-nutshell-15-320.jpg)
![What is a “new” drug
• The regulatory definition of "new drug“ is critical as it establishes
both the need for, and requirements for approval
A Ne • New Drug is an active substance which is not generally
recognized, among experts qualified to evaluate the safety and
effectiveness of drugs (FDA), as safe and effective for use under
the conditions prescribed, recommended, or suggested in the
labeling thereof
• Approved via IND/NDA
• Active compounds recognized under the Food and Drugs Act of
June 30, 1906 are the exception [“Old Drug”]
16](https://image.slidesharecdn.com/fdaapplicationsinanutshell-141021085414-conversion-gate01/85/FDA-applications-in-a-nutshell-16-320.jpg)