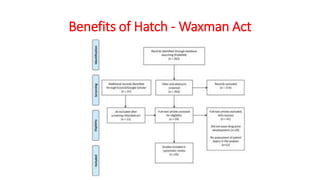
This document discusses generic drug product development. It defines generic drugs as having the same active ingredients, dosage, strength, quality and performance as brand name drugs. The main goals of generic drugs are to decrease drug prices after patents expire. The generic drug development process involves concept development, system design, detailed design, testing, and production ramp up. It also discusses the legislative history around generic drugs in the US, including the 1984 Hatch-Waxman Act, which established the Abbreviated New Drug Application pathway to expedite generic drug approval.