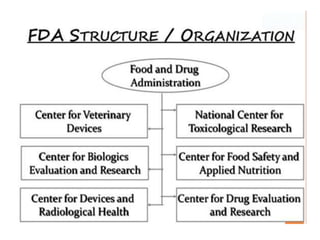
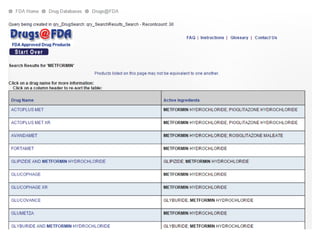
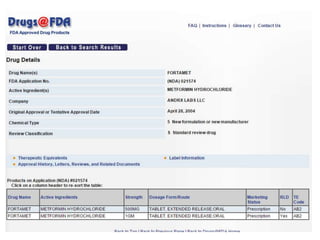
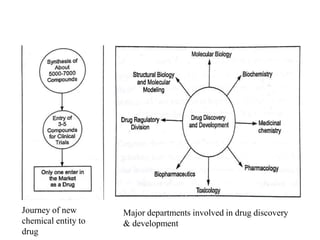
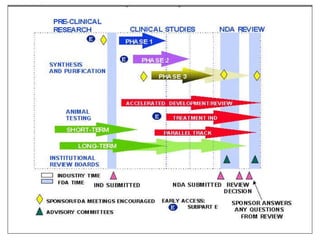
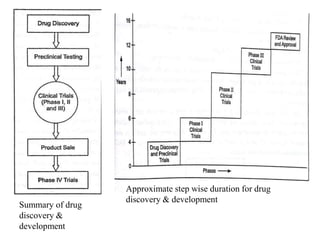
The document provides an overview of the drug development and approval process at the US Food and Drug Administration (FDA). It describes the FDA's structure and responsibilities in regulating drugs, medical devices, food, cosmetics, and other products. The key stages of drug development include drug discovery, pre-clinical testing, clinical trials (Phase I-III), and new drug application. FDA approval is required through this rigorous process to ensure safety and effectiveness before a new drug can reach consumers.
































































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)











