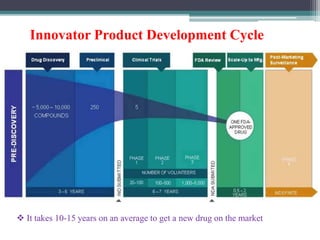
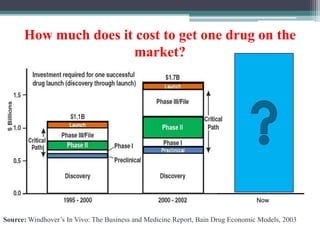
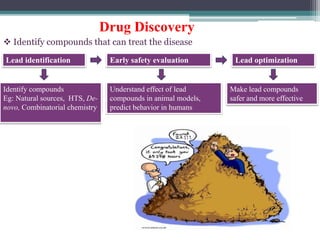
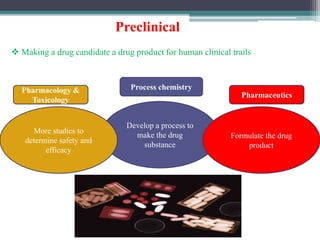
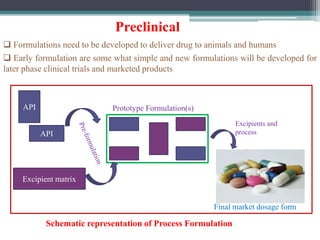
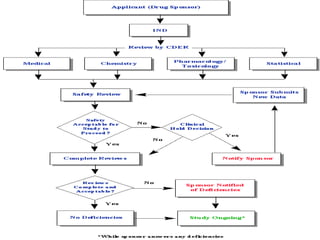
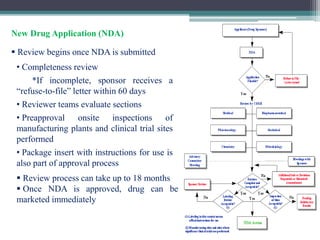
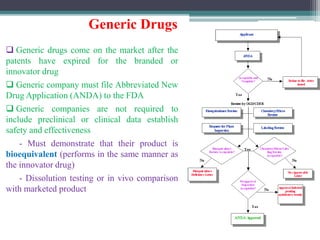
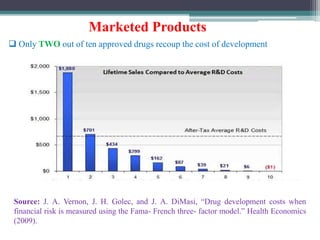
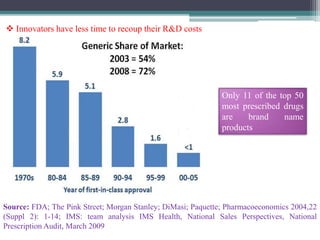
The document discusses the process for approval of a new drug from development through marketing. It takes 10-15 years on average and costs over $2.6 billion to get a new drug approved. Key steps include:
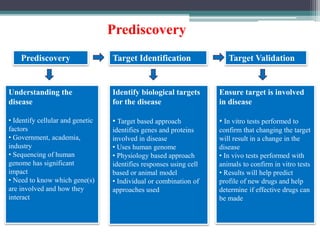
- Preclinical research to identify biological targets and compounds
- FDA approval to begin clinical trials in three phases involving thousands of subjects to test safety, efficacy, and dosing
- New Drug Application submission including all clinical trial data for FDA review and approval
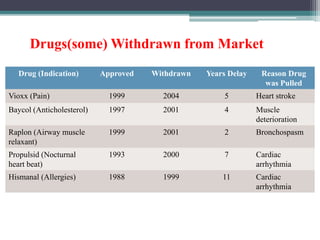
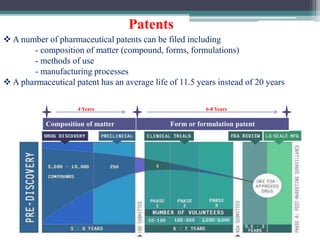
- Post-marketing studies and generic approval after patents expire