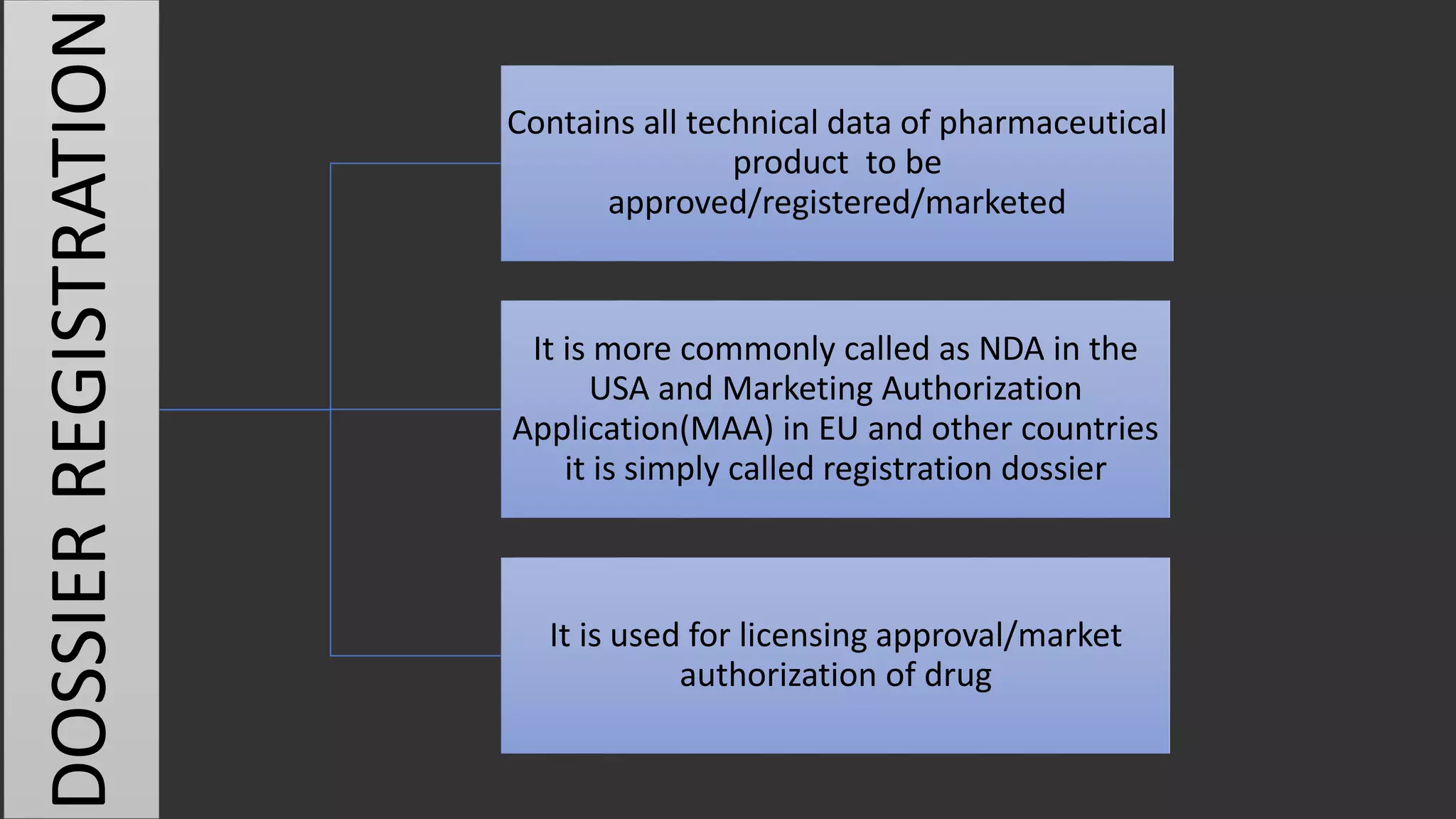
The document outlines the requirements and guidelines for submitting a drug product dossier in the CTD format as adopted by India’s CDSCO in alignment with ICH standards. It details the structure of the dossier, which includes administrative, quality, non-clinical, and clinical data necessary for regulatory approval. Moreover, it highlights the history and evolution of the CTD guidelines and the process for dossier preparation and submission.