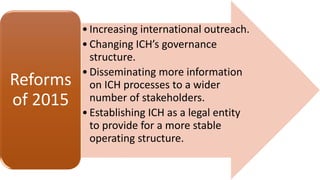
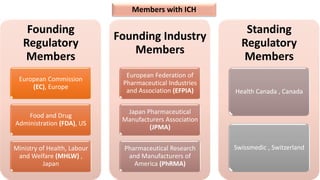
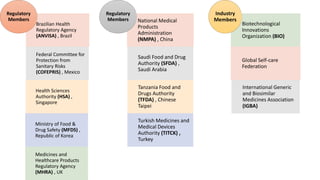
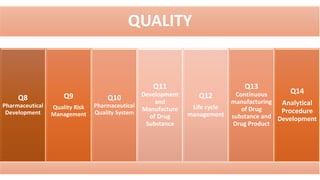
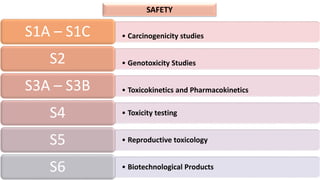
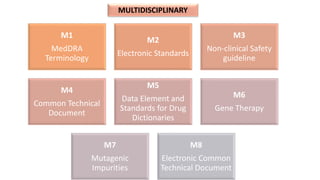
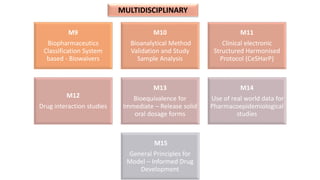
The document outlines the history, objectives, and guidelines of the International Council for Harmonisation (ICH), which was established in the 1990s to enhance global pharmaceutical regulation and promote safe, effective, and high-quality medicines. ICH's four pillars focus on quality, safety, efficacy, and multidisciplinary topics, ensuring harmonization among regulatory authorities and pharmaceutical industries in various countries. The document also details the membership structure, key guidelines, and recent reforms aimed at improving operational stability and information dissemination.