Downloaded 1,148 times








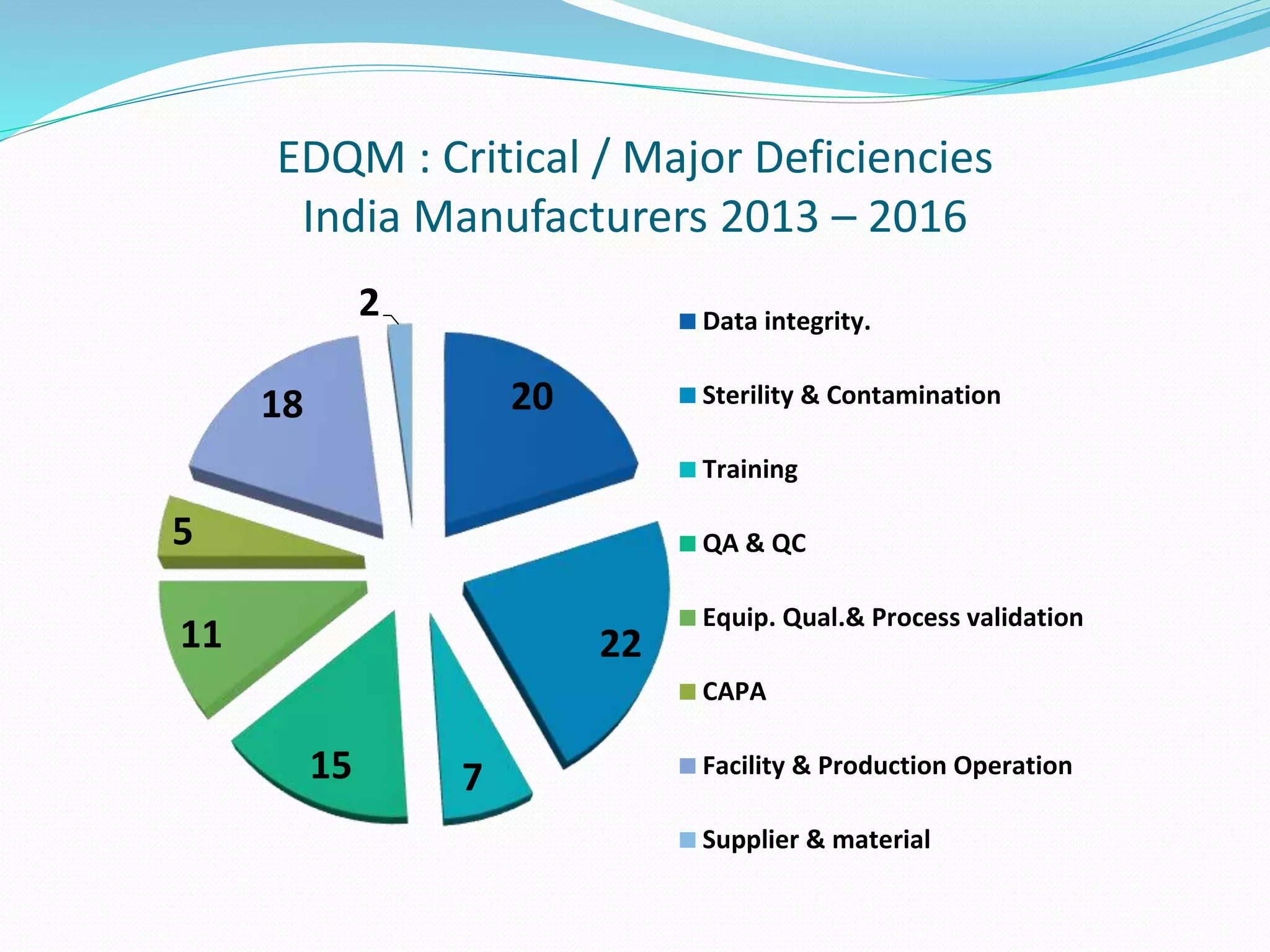






















This document discusses data integrity and its importance in the pharmaceutical industry. It defines data integrity as the extent to which all data is complete, consistent and accurate throughout its lifecycle. Regulatory agencies like the FDA and MHRA require data integrity to ensure patient safety. Lack of data integrity can result in warning letters or consent decrees from regulators. The document outlines principles like ALCOA+ to ensure data integrity. It discusses examples of data integrity issues found in warning letters and gives recommendations for proper implementation of data integrity controls and quality culture.























































































