Downloaded 147 times














































![Chemical cleaning
solutions
• For both organic and inorganic
contaminants
• General chemical cleaning
1. Sulphuric acid
• Hot sulphuric acid with added oxidant
• Also a general photoresist stripper
• H2SO4 is an effective cleaner in 90-125°C → remove
most inorganic residues and particulates from the
surface
• Oxidants are added to remove carbon residues
• Chemical reaction converts C to CO2
• Typical oxidants: hydrogen peroxide (H2O2),
ammonium persulfate [(NH4)2S2O8]
• Nitric acid (HNO3), and ozone (O2)](https://image.slidesharecdn.com/4-contaminationcontrol-21-150115102612-conversion-gate02/85/contamination-control-58-320.jpg)




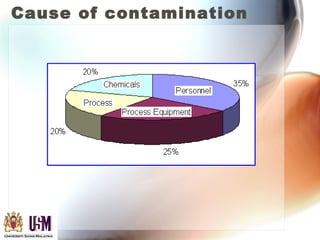
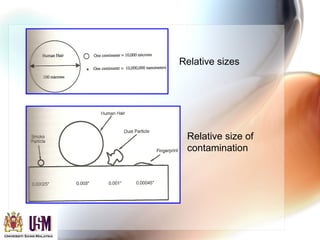
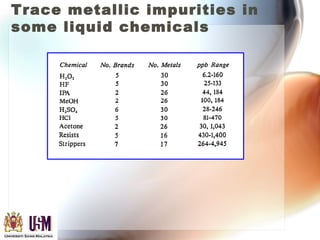
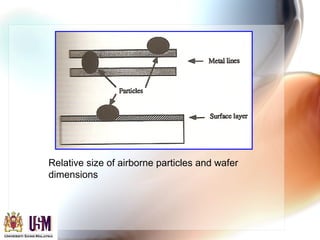
This document discusses contamination control in semiconductor fabrication. It identifies five major classes of contaminants: particles, metallic ions, chemicals, bacteria, and airborne molecular contaminants. Contamination can reduce device yield, affect performance and reliability. Sources of contamination include air, the production facility, cleanroom personnel, process water, chemicals, gases and static charge. The document describes techniques for maintaining cleanliness, such as cleanrooms, air filtration, garment protocols and chemical purification. It also outlines requirements for wafer cleaning and differences between front-end-of-line and back-end-of-line processing.























































































