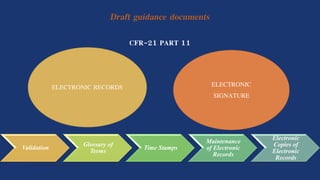
This document discusses CFR 21 Part 11, which outlines regulations set by the FDA for electronic records and electronic signatures to ensure their reliability and equivalence to traditional paper records. It includes details on general provisions, requirements for electronic records and signatures, and criteria for system validation and security. The goal of these regulations is to facilitate the use of electronic technology while maintaining public health protection.